PRMT5 modulates the metabolic response to fasting signals
Abstract
Under fasting conditions, increases in circulating glucagon maintain glucose balance by promoting hepatic gluconeogenesis. Triggering of the cAMP pathway stimulates gluconeogenic gene expression through the PKA-mediated phosphorylation of the cAMP response element binding (CREB) protein and via the dephosphorylation of the latent cytoplasmic CREB regulated transcriptional coactivator 2 (CRTC2). CREB and CRTC2 activities are increased in insulin resistance, in which they promote hyperglycemia because of constitutive induction of the gluconeogenic program. The extent to which CREB and CRTC2 are coordinately up-regulated in response to glucagon, however, remains unclear. Here we show that, following its activation, CRTC2 enhances CREB phosphorylation through an association with the protein arginine methyltransferase 5 (PRMT5). In turn, PRMT5 was found to stimulate CREB phosphorylation via increases in histone H3 Arg2 methylation that enhanced chromatin accessibility at gluconeogenic promoters. Because depletion of PRMT5 lowers hepatic glucose production and gluconeogenic gene expression, these results demonstrate how a chromatin-modifying enzyme regulates a metabolic program through epigenetic changes that impact the phosphorylation of a transcription factor in response to hormonal stimuli.
Chronic elevations in circulating blood glucose contribute to the chronic complications of type II diabetes, which include retinal blindness, renal failure, peripheral neuropathy, and cardiovascular diseases (1). Fasting hyperglycemia represents an early sign of insulin resistance, which is characterized by an inability for insulin to promote glucose uptake into muscle and to inhibit glucose production by the liver.
During fasting, increases in circulating pancreatic glucagon stimulate the gluconeogenic program via the protein kinase A (PKA)-mediated phosphorylation and activation of the transcription factor cAMP response element binding (CREB) protein, a modification that promotes recruitment of the coactivators P300 and CREB binding protein (CBP) (2). In parallel, increases in cAMP signaling also promote the dephosphorylation of the CREB regulated transcriptional coactivator 2 (CRTC2) (3). Sequestered in the cytoplasm through phosphorylation-dependent interactions with 14-3-3 proteins, CRTC2 undergoes dephosphorylation in response to cAMP, where it shuttles to the nucleus and mediates induction of cellular genes via an interaction with CREB over relevant binding sites (4, 5).
CRTC2 dephosphorylation represents an early event in the cAMP response relative to CREB phosphorylation, although the extent to which the two processes are linked remains unclear (6). The PKA-mediated phosphorylation of CREB on chromatin templates appears to be generally inefficient compared with its phosphorylation on naked DNA templates; these inhibitory effects can be circumvented through the acetylation of resident nucleosomes and subsequent increases in chromatin accessibility (7). The extent to which these or other chromatin modifications are permissive for CREB activation in vivo has not be addressed, however.
Here we characterize a CRTC2-associated arginine methylase that promotes CREB phosphorylation and target gene activation in part by enhancing chromatin accessibility over CREB binding sites. The results provide a mechanism to explain how latent cytoplasmic regulators such as CRTC2 may contribute to signaling in the nucleus through their association with chromatin modifying enzymes.
Results
In mass spectroscopy studies using epitope-tagged CRTC2 to identify relevant interacting proteins, we recovered the protein arginine methyltransferase 5 (PRMT5) (Fig. S1 A and B). We confirmed the CRTC2:PRMT5 interaction in coimmunoprecipitation studies with epitope-tagged proteins (Fig. 1A). The CRTC2:PRMT5 association was evident under basal conditions and to a greater extent following exposure to cAMP agonist forskolin (FSK).
Fig. 1.
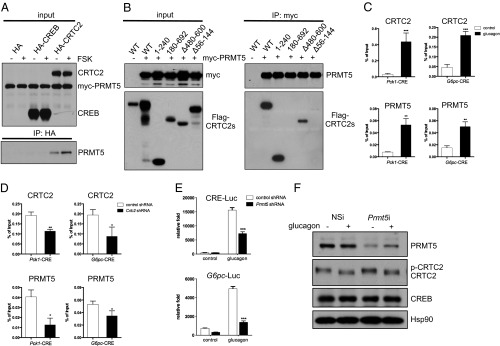
PRMT5 associates with CRTC2 and is recruited to gluconeogenic genes in response to glucagon. (A) Coimmunoprecipitation assay of HEK293T cells expressing epitope-tagged PRMT5 and CRTC2. No interaction with epitope-tagged CREB as shown. Exposure to FSK (30 min) indicated. (B) Coimmunoprecipitation assay of WT and mutant CRTC2 polypeptides with PRMT5 in cells exposed to FSK for 30 min. Amino acid endpoints for each construct indicated. (C) ChIP assay of primary hepatocytes showing effect of glucagon (1 h) on recruitment of PRMT5 and CRTC2 to CREB-binding sites over gluconeogenic (Pck1, G6pc) promoters. Each bar represents averaged results, n = 3 biological replicates, assayed three times each; error bars, SD. **P < 0.01; ***P < 0.001. (D) Effect of CRTC2 depletion by RNAi-mediated knockdown on PRMT5 recruitment to gluconeogenic promoters in primary hepatocytes exposed to glucagon (1 h). Each bar represents averaged results, n = 2 biological replicates, assayed three times each; error bars, SD. *P < 0.05; **P < 0.01. (E) Effect of PRMT5 depletion on CRE-Luc and G6pc-Luc reporter activities in primary hepatocytes exposed to glucagon (4–6 h). Each bar represents averaged results, n = 3 biological replicates, assayed three times each; error bars, SD. ***P < 0.001. (F) Western blot showing effect of Prmt5 RNAi on PRMT5 protein amounts and on CRTC2 dephosphorylation in primary hepatocytes exposed to glucagon (30 min). Hsp, heat shock protein 90.
CRTC2 and its paralogs share a similar structure consisting of an N-terminal CREB-binding domain (amino acids 1–55), central regulatory domain (amino acids 55–600), and C-terminal transactivation domain (amino acids 600–692) (2). In deletion mapping studies, PRMT5 was found to associate with the central regulatory region of CRTC2; deletion of sequences within this domain (amino acids 56–144) disrupted the CRTC2:PRMT5 interaction (Fig. 1B).
The PRMT family of arginine methylases consists of three types; they are distinguished by their ability to carry out mono- or asymmetric di-methylation (type I), mono- or symmetric di-methylation (type II), or exclusively monomethylation (type III) (8, 9). PRMT5 functions as the major type II symmetric arginine methyltransferase. Consistent with a critical role during development, mice with a knockout of Prmt5 have early embryonic lethality (10). PRMT5 has also been shown to function in transcriptional regulation as well as signal transduction (11).
Based on the ability for PRMT5 to associate with CRTC2, we tested whether this enzyme is correspondingly recruited to CREB target genes in response to fasting signals. In ChIP studies, exposure of mouse primary hepatocytes to glucagon increased amounts of CRTC2 over CREB-binding sites on the gluconeogenic glucose-6-phosphatase (G6pc) and phosphoenolpyruvate carboxykinase (Pck1) promoters (Fig. 1C). Consistent with its ability to associate with CRTC2, PRMT5 was also recruited to these CREB target genes following exposure to glucagon. Depletion of Crtc2 with adenovirally encoded RNAi disrupted PRMT5 recruitment in hepatocytes exposed to glucagon (Fig. 1D).
Supporting a functional role for PRMT5 in modulating CREB activity, exposure to glucagon increased cAMP response element (CRE)-luciferase (Luc) and G6pc-Luc reporter activities robustly; these effects were disrupted following RNAi-mediated depletion of Prmt5 (Fig. 1E). Conversely, overexpression of Crtc2 and Prmt5 increased G6pc-Luc reporter activity cooperatively in transient assays of HepG2 hepatoma cells (Fig. S1C). In keeping with the role of CRTC2 in promoting PRMT5 recruitment, PRMT5 had no effect on G6pc-Luc reporter activity in cells depleted of Crtc2 (Fig. S1D). Arguing against a direct effect on cAMP signaling per se, knockdown of Prmt5 did not interfere with the dephosphorylation and hence activation of CRTC2 in hepatocytes exposed to glucagon (Fig. 1F). Taken together, these results indicate that PRMT5 up-regulates CREB activity following its CRTC2-dependent recruitment to relevant genes in response to cAMP agonist.
We tested whether PRMT5 is required for induction of the gluconeogenic program during fasting. RNAi-mediated depletion of Prmt5 in liver decreased circulating glucose concentrations and gluconeogenic gene expression in lean mice and to a greater extent in genetically obese (ob/ob) or high-fat diet fed insulin-resistant mice (Fig. 2 A and B and Fig. S2 A–C). The effect of PRMT5 on hepatic gluconeogenesis appears cell autonomous because RNAi-mediated depletion of Prmt5 also down-regulated expression of Pck1, G6pc, and Peroxisome Proliferator Activated Receptor Gamma Coactivator 1a (Ppargc1a) genes in primary hepatocytes exposed to glucagon (Fig. 2C). As a consequence, knockdown of Prmt5 reduced glucose secretion from primary hepatocytes exposed to glucagon (Fig. 2D). Pointing to a specific effect on CREB signaling, Prmt5 knockdown did not alter mRNA amounts for insulin-like growth factor binding protein 1 (Igfbp1), a Forkhead box O1 (FOXO1) target gene that is also up-regulated during fasting (Fig. S2D).
Fig. 2.

PRMT5 stimulates hepatic gluconeogenesis during fasting and in diabetes. (A) Effect of hepatic Prmt5 depletion with adenovirally encoded RNAi on fasting blood glucose concentrations in genetically obese (ob/ob) mice compared with lean controls (ob/+). Each bar represents averaged results, n = 5; error bars, SD. **P < 0.01; ***P < 0.001. (B) Effect of hepatic PRMT5 knockdown on gluconeogenic gene expression in livers of fasted obese (ob/ob) mice. Each bar represents averaged results, n = 5; error bars, SD. **P < 0.01; ***P < 0.001. (C) Effect of PRMT5 knockdown on gluconeogenic gene expression in primary hepatocytes exposed to glucagon. Each bar represents averaged results, n = 3 biological replicates, assayed three times each; error bars, SD. *P < 0.05; **P < 0.01; ***P < 0.001. (D) Effect of PRMT5 depletion on glucose secretion from primary hepatocytes exposed to glucagon. Each bar represents averaged results, n = 3; error bars, SD. **P < 0.01. (E) Gene profiling analysis of primary mouse hepatocytes following RNAi-mediated knockdown of Prmt5. Pie charts show proportion of total cellular genes up or down-regulated 1.5-fold or better under basal conditions and following exposure to glucagon. (F) Heat map showing effects of Prmt5 knockdown on top 40 scoring glucagon-inducible genes in primary hepatocytes.
PRMT5 has been reported to exert both positive and negative effects on cellular gene expression (8), prompting us to evaluate relative effects of this methylase in primary hepatocytes by gene profiling analysis. Depletion of Prmt5 down-regulated the expression of 1.5% of cellular genes, whereas it up-regulated the expression of about 1% of genes (Fig. 2E). By contrast, Prmt5 depletion decreased the expression of 20% of glucagon-inducible genes, many of which are direct targets for CREB that function in fasting glucose metabolism [tyrosine amino transferase (Tat), G6pc, Pck1, Ppargc1a (Fig. 2F). Consistent with its proposed role in potentiating CREB activity, knockdown of Prmt5 did not up-regulate the expression of any glucagon-inducible gene in this setting.
PRMT5 has been shown to catalyze the symmetric di-methylation of histone H3 at Arg-2 (H3R2me2-s) and at Arg-8 (H3R8me2-s); these marks have been associated with transcriptional repression as well as activation (8, 12). In turn, H3R2me2-s has been reported to modulate cellular gene expression in part by promoting the recruitment of WD repeat protein 5 (WDR5) (12), a core subunit of lysine methyl transferase complexes (13) and a component of the transcriptional adaptor (Ada) 2A containing complex (ATAC) (14). We confirmed that PRMT5 catalyzes increases in H3R2me2-s in vitro and that H3R2me2-s in turn associates with WDR5 by GST pull-down assays (Fig. 3 A and B).
Fig. 3.

PRMT5 promotes H3R2 symmetric dimethylation and recruitment of WDR5 to gluconeogenic promoters in response to glucagon. (A) Western blot showing effect of PRMTs (5–7) on H3R2me2 and H3R2me2-s of histone H3 Arg-2 in vitro. (B) Western blot showing effect of PRMT5 and PRMT6-catalyzed histone H3 Arg-2 di-methylation on WDR5 binding by pull-down assay with GST-WDR5. (C) ChIP assay of primary hepatocytes, showing effect of glucagon (1 h) on H3R2me2-s methylation and WDR5 recruitment at CREB-binding sites over gluconeogenic promoters. Each bar represents averaged results, n = 3 biological replicates, assayed three times each; error bars, SD. *P < 0.05; **P < 0.01. (D) ChIP assay showing effect of Prmt5 knockdown on H3R2me2-s and WDR5 amounts over gluconeogenic promoters in hepatocytes exposed to glucagon (1 h). Each bar represents averaged results, n = 3 biological replicates, assayed three times each; error bars, SD. *P < 0.05; **P < 0.01.
Consistent with these observations, exposure of primary hepatocytes to glucagon increased H3R2me2-s amounts alongside the active mark histone H3 trimethyl-lysine 4 at CREB-binding sites over gluconeogenic genes (Fig. 3C and Fig. S3A). In keeping with these increases, exposure to glucagon stimulated WDR5 recruitment to gluconeogenic promoters (Fig. 3C). Conversely, Prmt5 depletion decreased both H3R2me2-s and WDR5 amounts over these genes in primary hepatocytes following glucagon treatment (Fig. 3D). Depletion of Crtc2 also decreased H3R2me2-s amounts at Pck1 and G6pc promoters, demonstrating the importance of CRTC2 in mediating PRMT5 recruitment (Fig. S3B).
Based on the ability for PRMT5 to increase H3R2me2-s amounts over CREB target genes, we considered whether this methylase could open chromatin structure at relevant promoters in response to glucagon. Supporting this idea, exposure to glucagon decreased nucleosome occupancy over CREB-binding sites in primary hepatocytes as revealed by decreases in histone H3 occupancy; these effects were blocked following RNAi-mediated knockdown of Prmt5 or Crtc2 (Fig. 4 A and B and Fig. S4A). Taken together, these results indicate that PRMT5 enhances CREB activation in part by increasing H3R2me2-s amounts over CREB binding sites in response to cAMP agonist.
Fig. 4.
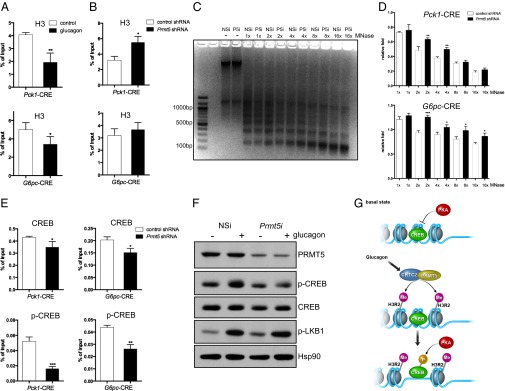
PRMT5 promotes CREB phosphorylation at gluconeogenic promoters by increasing chromatin accessibility at CREB-binding sites. (A) Effect of glucagon (1 h) exposure on total histone H3 amounts over CREB-binding sites on gluconeogenic genes in primary hepatocytes. Each bar represents averaged results, n = 3 biological replicates, assayed three times each; error bars, SD. *P < 0.05; **P < 0.01. (B) Effect of Prmt5 knockdown on H3 occupancy over CREB-binding sites at gluconeogenic promoters in hepatocytes exposed to glucagon for 1 h. Each bar represents averaged results, n = 3 biological replicates, assayed three times each; error bars, SD. *P < 0.05. (C) Effect of PRMT5 depletion on MNase digestion of chromatin from primary hepatocytes exposed to glucagon (1 h). Increasing amounts of MNase indicated. (D) Effect of Prmt5 RNAi on MNase sensitivity over CREB-binding sites on Pck1 and G6pc promoters in cells exposed to glucagon (1 h). Representative experiment from three independent experiments shown. Each bar represents averaged results, assayed three times each; error bars, SD. *P < 0.05; **P < 0.01. ***P < 0.001. (E) ChIP assay showing effect of Prmt5 RNAi on CREB and p-CREB amounts over gluconeogenic promoters in primary hepatocytes exposed to glucagon (1 h). Each bar represents averaged results, n = 3 biological replicates, assayed three times each; error bars, SD. *P < 0.05; **P < 0.01. ***P < 0.001. (F) Western blot showing effect of PRMT5 depletion on amounts of p-CREB and total CREB in primary hepatocytes under basal conditions and following exposure to glucagon (30 min). Relative phosphorylation of LKB in response to glucagon shown for comparison. (G) Model for activation of gluconeogenic genes during fasting. Exposure to glucagon leads to CRTC2 activation and association with PRMT5. Following its CRTC2-mediated recruitment to relevant target genes, PRMT5 increases nucleosome clearance over CREB-binding sites, in part through increases in histone H3 Arg2 di-methylation that promote WDR5 occupancy. As a consequence of the increase in chromatin accessibility, CREB phosphorylation by PKA is up-regulated over PRMT5 occupied promoters, leading to increases in target gene expression. NSi, nonspecific RNAi; P5i, PRMT5 RNAi; p-LKB1, phospho-liver kinase B1.
We reasoned that PRMT5 may enhance chromatin accessibility over CREB-regulated promoters. To test this idea, we evaluated effects of PRMT5 on micrococcal nuclease (MNase) sensitivity in control and Prmt5-depleted cells in response to glucagon. Although it did not alter MNase sensitivity globally, Prmt5 depletion blocked glucagon-dependent increases in MNase sensitivity over G6pc and Pck1 promoters (Fig. 4 C and D). As a result, Ser133-phosphorylated CREB (p-CREB) amounts over gluconeogenic genes were substantially reduced in Prmt5-depleted compared with control cells exposed to glucagon (Fig. 4E and Fig. S4B). Similarly, depletion of Crtc2 reduced p-CREB amounts over gluconeogenic promoters in cells exposed to glucagon, reflecting in part the importance of this coactivator in mediating PRMT5 recruitment (Fig. S4C). Although Prmt5 depletion decreased p-CREB amounts in cells exposed to glucagon, it did not interfere with the phosphorylation of cytosolic PKA substrates such as liver kinase B1 (LKB1) (Fig. 4F). Collectively, these results demonstrate that PRMT5 modulates the expression of signal-dependent genes through increases in chromatin accessibility that are permissive for the phosphorylation of nuclear factors such as CREB by PKA and perhaps other regulatory enzymes (Fig. 4G).
Discussion
Under fasting conditions, increases in circulating glucagon promote hepatic glucose production via the cAMP-dependent phosphorylation of CREB and dephosphorylation of CRTC2 (2). CREB and CRTC2 activities are increased in diabetes, where they contribute to the attendant hyperglycemia via up-regulation of the gluconeogenic program.
We found that CRTC2 stimulates gluconeogenic gene expression via an association with the protein arginine methyltransferase PRMT5 in response to glucagon stimulation. Following its recruitment to CREB-binding sites, PRMT5 promotes increases in H3R2me2-s that in turn trigger the expression of relevant CREB target genes. PRMT5-dependent increases in H3R2me2-s appeared to stimulate cellular gene expression in part by enhancing the recruitment of WDR5. The CRTC2-dependent recruitment of this chromatin-modifying enzyme to CREB-binding sites appears critical for the subsequent phosphorylation of CREB.
In addition to effects of PRMT5, the type I PRMT family member PRMT4/coactivator-associated arginine methyltransferase 1 (CARM1) has also been found to promote gluconeogenic gene expression via an interaction with CREB and through subsequent increases in H3 methylation (15). Indeed, we have also detected increases in asymmetric H3R8me2 amounts at CREB-binding sites, potentially catalyzed by the type I enzyme PRMT2, in response to glucagon simulation. Collectively, these studies indicate that histone arginine methylation plays an important role in hepatic gluconeogenesis.
Phosphorylation has been shown to modulate transcription factors and coactivators in the cytoplasm by regulating their nuclear translocation, and in the nucleus by increasing their transactivation potential (16). Consistent with its proximity to the signaling machinery in the cytoplasm, CRTC2 undergoes rapid and quantitative dephosphorylation in response to glucagon. Although the two events are closely spaced, CRTC2 dephosphorylation and promoter recruitment appear to precede cAMP-dependent increases in phospho-CREB levels, suggesting a potential role for CRTC2 in contributing to CREB activation (6).
In reconstitution studies, CREB phosphorylation by PKA was strongly inhibited on chromatin compared with naked DNA templates, whereas CREB occupancy was more resistant (7). We found that CRTC2 enhances CREB phosphorylation in response to glucagon via its association with PRMT5. In turn, PRMT5 potentiated CREB phosphorylation through increases in H3R2me2-s that stimulate nucleosome clearance and thereby increase accessibility to PKA. Taken together, these results indicate that local changes in chromatin structure may be required for signaling to certain nuclear factors. Future studies should reveal whether such changes are predictive for the selective activation of target gene subsets by CREB and other phosphorylation-dependent factors in response to extracellular stimuli.
Materials and Methods
Cell Culture.
HEK293T cells were cultured in DMEM with 10% (wt/vol) FBS and 1× Pen/Strep at 37 °C in 5% (wt/vol) CO2 and exposed to FSK (10 μM). Mouse primary hepatocytes were isolated as previously described and cultured in Medium 199 and exposed to glucagon (100 nM) (18).
Coimmunoprecipitation.
HEK293T cells and cultured mouse primary hepatocytes were plated in six-well plates with or without FSK or glucagon treatment. HEK293T cells were transfected with indicated plasmids using Effectene (Qiagen) following manufacturer’s instructions. Typically, 1-μg plasmids were used for each transfection. Protein lysate was collected 24 h after transfection. The treated cells were lysed in NTDT buffer (150 mM NaCl, 50 mM Tris⋅HCl, 0.1% deoxycholate, and 1% TX-100). The cellular debris was pelleted by centrifugation and the supernatant (immunoprecipitation lysate) was collected and diluted for coimmunoprecipitation experiments. The immunoprecipitation lysate was precleared with 20 μL protein A beads for 1 h at 4 °C. Ethidium bromide was added to a final concentration of 100 μg/mL to prevent protein–DNA binding. Precleared, ethidium bromide-treated lysate was incubated with indicated antibody-bound agarose beads overnight at 4 °C. Protein/antibody-bound beads were recovered by centrifugation and washed three times with NTP buffer (150 mM NaCl, 50 mM Tris⋅HCl, 0.1% Nonidet P-40). The protein/antibody-bound beads were incubated with 3× SDS/PAGE loading buffer for 5 min at 95 °C and subjected to SDS/PAGE.
Mass spectroscopy studies were performed on precipitates from Halo-tagged CRTC2 as reported (19).
Chromatin Immunoprecipitation Assay.
Cultured mouse primary hepatocytes were plated in 150-mm plates and exposed to glucagon for 1 h. Adenovirally encoded shRNAs were added to cells for 60 h. ChIP assays were performed as described, with minimal modification (20). Briefly, the treated hepatocytes were cross-linked with 1% formaldehyde for 15 min. Cross-linking reactions were stopped with 0.125 M glycine. Cross-linked cells were washed by PBS three times and stored at −80 °C before use. Fragmented, precleared chromatin lysate was incubated overnight with indicated antibodies: H3, histone H3 trimethyl-lysine 4, and PRMT5 (Active Motif), PRMT5, and WDR5 (Millipore).
Luciferase Reporter Assay.
Cultured mouse primary hepatocytes were infected with Ad-CRE-Luc or Ad-G6PC-Luc and adenovirally encoded shRNAs for 60 h and exposed to glucagon for 4–6 h. Luciferase assays were performed as described (21).
Mouse Strains and in Vivo Analyses.
High-fat diet-fed ob/+ and ob/ob mice were purchased from Jackson Laboratories. All mice were adapted to their environment for 1 wk before study. Adenovirus encoded with indicated shRNAs (3 × 108 pfu) were delivered to mice by eye injection. Six days after infection, mice were fasted for 16 h, blood glucose concentrations were measured before and after fasting, and liver tissue was collected after fasting for analysis. Animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at the Salk Institute.
RNA Analyses.
Cellular RNA was isolated by RNeasy kit (Qiagen) from mouse liver tissue or from mouse primary hepatocytes. Mouse primary hepatocytes were plated in six-well plates and infected with adenovirus encoded with indicated shRNAs for 60 h and exposed to glucagon for 1 and 2 h. mRNA levels were measured as described (20).
Glucose Secretion Analyses.
Mouse primary hepatocytes were plated in six-well plates and exposed to glucagon for 7 h. Glucose concentrations in conditioned medium were measured as described (22).
Gene Profiling Analyses.
The mouse primary hepatocytes were plated in 60-mm plates and infected with adenovirus encoded with indicated shRNAs for 60 h and exposed to glucagon for 1.5 h. RNA was isolated and analyzed by Affymetrix GeneChip Mouse Gene 1.0 ST Array following manufacturer’s instructions. Top 40 glucagon-induced genes were chosen for heat map analysis using matrix2png software (23).
In Vitro Methylation and GST Pull-Down Assays.
Recombinant H3 proteins (Active Motif) were methylated in vitro using purified PRMTs expressed either in bacteria as GST fusion proteins (GST-PRMT6, GST-PRMT7) or as myelocytomatosis (myc)-tagged proteins (myc-PRMT5) purified from HEK293T cells as described (24). Methylated H3 proteins were then isolated and incubated with GST-WDR5 proteins in NTP buffer (150 mM NaCl, 50 mM Tris⋅HCl, 0.5% Nonidet P-40) overnight. GST Sepharose beads were added to protein samples for 1 h and washed three times. The GST-WDR5/H3 bound beads were analyzed by SDS/PAGE.
MNase Digestion Analyses.
Mouse primary hepatocytes were plated in six-well plates and infected with adenovirus encoded with indicated shRNAs for 60 h and exposed to glucagon for 1 h. Micrococcal nuclease (MNase) digestion analyses were performed as previously described (25), with minimal modification. Briefly, the treated hepatocytes were permeabilized using 0.05% lysolecithin for 5 min, and treated with different amounts of MNase for 5min. DNA was purified and analyzed by quantitative PCR.
Statistical Analyses.
GraphPad Prism6 software (GraphPad Software, Inc.) was used for analysis of P values based on at least two independent experiments in three independent PCR reactions. The two-tailed unpaired t test was used to compare the differences between two groups. P values < 0.05 were considered statistically significant, *P < 0.05, **P < 0.01, and ***P < 0.001.
Supplementary Material
Supporting Information
Acknowledgments
We thank M. Bedford for PRMT plasmids and K. Ravnskjaer and J. Butler for advice and reagents. Funding for this work was provided by National Institutes of Health Grants F32 DK096778 (to W.-W.T.), R01 DK049777, R01 DK083834, and R01 DK091618; the Clayton Foundation for Medical Research; the Leona M. and Harry B. Helmsley Charitable Trust; and the Kieckefer Foundation.
Footnotes
The authors declare no conflict of interest.
References
- 1.Saltiel AR. New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell. 2001;104(4):517–529. doi: 10.1016/s0092-8674(01)00239-2. [DOI] [PubMed] [Google Scholar]
- 2.Altarejos JY, Montminy M. CREB and the CRTC co-activators: Sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011;12(3):141–151. doi: 10.1038/nrm3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koo SH, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437(7062):1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- 4.Screaton RA, et al. The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell. 2004;119(1):61–74. doi: 10.1016/j.cell.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 5.Bittinger MA, et al. Activation of cAMP response element-mediated gene expression by regulated nuclear transport of TORC proteins. Curr Biol. 2004;14(23):2156–2161. doi: 10.1016/j.cub.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Ravnskjaer K, et al. Cooperative interactions between CBP and TORC2 confer selectivity to CREB target gene expression. EMBO J. 2007;26(12):2880–2889. doi: 10.1038/sj.emboj.7601715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michael LF, Asahara H, Shulman AI, Kraus WL, Montminy M. The phosphorylation status of a cyclic AMP-responsive activator is modulated via a chromatin-dependent mechanism. Mol Cell Biol. 2000;20(5):1596–1603. doi: 10.1128/mcb.20.5.1596-1603.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bedford MT, Clarke SG. Protein arginine methylation in mammals: Who, what, and why. Mol Cell. 2009;33(1):1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bedford MT, Richard S. Arginine methylation an emerging regulator of protein function. Mol Cell. 2005;18(3):263–272. doi: 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 10.Tee WW, et al. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 2010;24(24):2772–2777. doi: 10.1101/gad.606110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bao X, et al. Overexpression PRMT5 promotes tumor cell growth and is associated with poor disease prognosis in epithelial ovarian cancer. J Histochem Cytochem. 2013;61(3):206–217. doi: 10.1369/0022155413475452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Migliori V, et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat Struct Mol Biol. 2012;19(2):136–144. doi: 10.1038/nsmb.2209. [DOI] [PubMed] [Google Scholar]
- 13.Wysocka J, et al. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell. 2005;121(6):859–872. doi: 10.1016/j.cell.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 14.Krebs AR, Karmodiya K, Lindahl-Allen M, Struhl K, Tora L. SAGA and ATAC histone acetyl transferase complexes regulate distinct sets of genes and ATAC defines a class of p300-independent enhancers. Mol Cell. 2011;44(3):410–423. doi: 10.1016/j.molcel.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krones-Herzig A, et al. Signal-dependent control of gluconeogenic key enzyme genes through coactivator-associated arginine methyltransferase 1. J Biol Chem. 2006;281(6):3025–3029. doi: 10.1074/jbc.M509770200. [DOI] [PubMed] [Google Scholar]
- 16.Hunter T, Karin M. The regulation of transcription by phosphorylation. Cell. 1992;70(3):375–387. doi: 10.1016/0092-8674(92)90162-6. [DOI] [PubMed] [Google Scholar]
- 17.Mayall TP, Sheridan PL, Montminy MR, Jones KA. Distinct roles for P-CREB and LEF-1 in TCR alpha enhancer assembly and activation on chromatin templates in vitro. Genes Dev. 1997;11(7):887–899. doi: 10.1101/gad.11.7.887. [DOI] [PubMed] [Google Scholar]
- 18.Dentin R, et al. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature. 2007;449(7160):366–369. doi: 10.1038/nature06128. [DOI] [PubMed] [Google Scholar]
- 19.Qi L, et al. TRB3 links the E3 ubiquitin ligase COP1 to lipid metabolism. Science. 2006;312(5781):1763–1766. doi: 10.1126/science.1123374. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, et al. Targeted disruption of the CREB coactivator Crtc2 increases insulin sensitivity. Proc Natl Acad Sci USA. 2010;107(7):3087–3092. doi: 10.1073/pnas.0914897107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 2008;456(7219):269–273. doi: 10.1038/nature07349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, et al. Inositol-1,4,5-trisphosphate receptor regulates hepatic gluconeogenesis in fasting and diabetes. Nature. 2012;485(7396):128–132. doi: 10.1038/nature10988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pavlidis P, Noble WS. Matrix2png: A utility for visualizing matrix data. Bioinformatics. 2003;19(2):295–296. doi: 10.1093/bioinformatics/19.2.295. [DOI] [PubMed] [Google Scholar]
- 24.Butler JS, Zurita-Lopez CI, Clarke SG, Bedford MT, Dent SY. Protein-arginine methyltransferase 1 (PRMT1) methylates Ash2L, a shared component of mammalian histone H3K4 methyltransferase complexes. J Biol Chem. 2011;286(14):12234–12244. doi: 10.1074/jbc.M110.202416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamasaki T, et al. 2007. Assays of nucleosome assembly and the inhibition of histone acetyltransferase activity. (11) Digestion of chromatin; and (12) purification and characterization of DNA after digestion of chromatin. Protocol Exchange, 10.1038/nprot.2007.340.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information