Severe Human Lower Respiratory Tract Illness Caused by Respiratory Syncytial Virus and Influenza Virus Is Characterized by the Absence of Pulmonary Cytotoxic Lymphocyte Responses
Abstract
Background. Respiratory syncytial virus (RSV) and influenza virus are common causes of infantile lower respiratory tract infection (LRTI). It is widely believed that both viral replication and inappropriately enhanced immune responses contribute to disease severity. In infants, RSV LRTI is known to be more severe than influenza virus LRTI.
Methods. We compared cytokines and chemokines in secretions of infants surviving various forms of respiratory illness caused by RSV or influenza viruses, to determine which mediators were associated with more-severe illness. We analyzed lung tissue from infants with fatal cases of RSV and influenza virus LRTI to determine the types of inflammatory cells present. Autopsy tissues were studied for the lymphotoxin granzyme and the apoptosis marker caspase 3.
Results. Quantities of lymphocyte-derived cytokines were minimal in secretions from infants with RSV infection. Concentrations of most cytokines were greater in influenza virus, rather than RSV, infection. Lung tissues from infants with fatal RSV and influenza virus LRTI demonstrated an extensive presence of viral antigen and a near absence of CD8-positive lymphocytes and natural killer cells, with marked expression of markers of apoptosis.
Conclusions. Severe infantile RSV and influenza virus LRTI is characterized by inadequate (rather than excessive) adaptive immune responses, robust viral replication, and apoptotic crisis.
Infection with respiratory syncytial virus (RSV) is the most frequent cause of hospitalization of infants in the United States [1, 2]. The pathogenesis of illness related to RSV infection is not well understood. Recent articles by respected authorities in the field state that both viral replication in the lungs and vigorous immune responses (particularly lymphocyte responses) against RSV are critical elements in the pathogenesis of lower respiratory tract infection (LRTI) [3–11].
The belief that immune responses contribute to the pathogenesis of RSV LRTI derives largely from experiences with a formalin-inactivated RSV vaccine (FIRSV) preparation. Vaccine recipients developed greater lymphoproliferative responses to RSV antigen and more-severe forms of respiratory tract illness when subsequently infected with RSV than did control subjects [12, 13]. However, analysis of lung tissue from 2 vaccine recipients who later died of RSV infection showed eosinophilia and lymphocytic infiltration that are not characteristic of naturally occurring RSV LRTI [14, 15]. Thus, disease after FI-RSV administration, while possibly mediated by lymphocytes, appears not be identical to that of natural RSV infection.
Immunocompromised subjects may develop prolonged viral replication, clinical evidence of pneumonia with less-frequent and less-prominent wheezing, and alveolar infiltrates when infected with RSV [16, 17], whereas subjects with intact immune responses develop bronchiolar infiltrates and wheezing [18–20]. However, it has not been demonstrated that altered T lymphocyte function (as opposed to that of other cells) specifically accounts for these differences.
Finally, in mouse models, ablation of either CD4- or CD8- positive lymphocytes after RSV infection lessens the severity of illness [9]. Nevertheless, the mouse model lacks many of the pathological features of RSV infection in human infants, including the predominance of neutrophils in airway exudates [21, 22], the extensive bronchiolar epithelial destruction, and the plugging of small airways with inflammatory debris [14, 15, 18]. The pattern of viral replication may also differ [23, 24].
Therefore, although increased activity of some component of the inflammatory response to RSV infection may be critical to the development of severe forms of bronchiolitis, the nature of the overly responsive mediator remains unknown. In human infants, RSV generally causes a more-severe form of LRTI than does influenza virus [25, 26]. We therefore evaluated infants with LRTI caused by these 2 viruses for differences in the nature of the inflammatory response that might explain the greater severity of illness in subjects with RSV infection and elucidate the pathogenesis of RSV infection in infants.
Patients and Methods
Study populations. Subjects of the present study were from 2 population groups. One group consisted of surviving infants <12 months of age with either RSV or influenza virus infection recruited from inpatient and outpatient areas of the Women and Children's Hospital of Buffalo. These subjects were evaluated for cytokine content of nasopharyngeal secretions (NPS; table 1) (for details, see the Appendix, which appears only in the electronic edition of the Journal). These subjects were assigned a diagnosis of either upper respiratory tract illness (URTI) alone or bronchiolitis on the basis of criteria described elsewhere [27] and in the Appendix.
Table 1.

Demographic and clinical features of the study subjects.
A second group of infants had fatal LRTI. Postmortem lung tissue was obtained from infants with fatal RSV (n = 9) or influenza virus (n = 11) LRTI who were autopsied at Hospital Roberto del Río, Santiago, Chile. Features of these 20 infants are also described in table 1 and the Appendix. Two pathologists (L.V. and L.M.) independently judged that the cause of death in all 20 infants was severe LRTI, with typical sloughing of bronchiolar epithelium, plugging of the terminal bronchioles, and infiltration of the airway wall and of the alveoli macrophages and neutrophils. There was no histological evidence of bacterial infection of the lungs. Dying infants had not been subjected to prolonged mechanical ventilation or to the use of anti-inflammatory agents or antivirals.
Measurement of cytokines in NPS. NPS from subjects recruited for cytokine analysis were tested using the Bio-Plex Human Cytokine 17-Plex panel (Bio-Rad Laboratories). The panel includes interleukin (IL)-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12 (p70), IL-13, IL-17, granulocyte colony stimulating factor (G-CSF), granulocyte-macrophage (GM)—CSF, interferon (IFN)—g, monocyte chemoattractant protein (MCP)—1 (referred to as CCL2), macrophage inflammatory protein (MIP)—1β (referred to as CCL4), and tumor necrosis factor (TNF)—α. The lower limit of detection of mediators was 7.5 pg/mL.
Routine pathological and immunohistochemical (IHC) analysis. Lung tissue samples from infants with fatal LRTI and control tissues were processed for routine staining and for IHC staining (for details, see the Appendix).
Statistical analysis. Statistical analysis was done using the StatView program (version 5.0.2; SAS) as described in the Appendix. Logarithmic transformation was used to obtain normal distributions of data. In the figures and tables, data are presented as the mean ± SE of the log10 values for individual chemokines.
Results
Demographic and Clinical Factors
An analysis of demographic and clinical features of the subjects in whom cytokine analysis was completed is presented in the upper part of table 1. Subjects with RSV LRTI (bronchiolitis) had longer hospital stays and lower oxygen saturations at the time of sampling than did subjects with influenza virus bronchiolitis. Other factors were similar between groups.
Demographic features of infants who died of LRTI are presented in the upper part of table 1. These factors were similar to those for subjects who survived, except that subjects with congenital heart disease and Down syndrome were not excluded. Preexisting conditions present before the onset of LRTI in these infants are listed the lower part table 1.
Frequency of Detection of Mediators
IL-2, IFN-γ, and IL-17. IL-2 was detectable in secretions from only 2 (5.6%) of 36 subjects with RSV infection but was detectable in secretions from 8 (24.2%) of 33 subjects with influenza virus infection (P = .04, data not shown). IFN-γ was detected in 13 (36.1%) of 36 of subjects with RSV infection and in 32 (97%) of 33 subjects with influenza virus infection (P < .0001). IL-17 was undetectable in subjects with RSV infection but was detected in 19 (57.6%) of 33 subjects with influenza virus infection (P < .0001).
IL-4, IL-5, and IL-13. IL-4 was detected in secretions from only 6 (16.7%) of 36 subjects with RSV infection and was detected in secretions from 21 (61.6%) of 33 of subjects with influenza virus infection (P = .0001). IL-5 and IL-13 were found infrequently (range, 0%–24%) in subjects with RSV or influenza virus infection, with no statistically significant differences between groups (P ⩾ .23, for each).
IL-12, MCP-1, and IL-6. IL-12 (P = .04), MCP-1 (P = .047), and IL-6 (P = .015) were detected less frequently in subjects with RSV infection than in those with influenza virus infection.
IL-1b, IL-7, and IL-10. IL-1β (P < .0001), IL-7 (P < .0001), and IL-10 (P = .0025) were all less frequently detected in subjects with RSV infection than in those with influenza virus infection.
G-CSF, GM-CSF, TNF-α, IL-8, and MIP-1β. G-CSF, GMCSF, TNF-α, IL-8, and MIP-1β were detected at frequencies that did not differ markedly (P ⩾ .24) between subjects with RSV or influenza virus infection.
Therefore, most cytokines and chemokines were detected more frequently among infants with influenza virus infection, and, surprisingly, none were found more commonly in those with RSV infection.
Concentrations of Mediators in Secretions and Type of Virus Infection
We next evaluated the measured quantities of mediators in secretions. First we compared cytokine responses between all subjects with RSV infection and those with influenza virus infection (figure 1A–1F). Quantities of most cytokines were greater in subjects with influenza virus infection than in subjects with RSV infection. These included cytokines presumed to be released predominantly from lymphocytes, such as IFN-γ and IL-17 (figure 1A). IL-4 (also predominantly released by lymphocytes) was similarly found in higher concentrations in infants with influenza virus infection, whereas other Th2 cytokines (IL-5 and IL-13) were either absent or present only in low concentrations in subjects with either type of virus infection (mean values at the lower limit of detection; figure 1B). Quantities of IL-12, MCP-1, and IL-6 (prominently produced by macrophages, among other cells) and of IL-1β, IL-7, and IL-10 (various cell types) were of a greater magnitude in infants with influenza virus infection than in those with RSV infection, particularly those with influenza bronchiolitis (figure 1C and 1D). Concentrations of G-CSF, GM-CSF, TNF-α, IL-8, and MIP-1β (notably released from epithelial cells, among others) were present at high concentrations and were not significantly different in subjects with either type of virus infection (figure 1E and 1F).
Figure 1.

Quantities of cytokines and chemokines in nasopharyngeal secretions of infants surviving respiratory syncytial virus (RSV) or influenza virus infection. Concentrations (log10) of each cytokine are illustrated for subjects with upper respiratory tract illness alone or bronchiolitis caused by each virus. The horizontal bars within boxes indicate the 50th percentile, the limits of boxes indicate the 25th and 75th percentiles, and the vertical lines indicate the third and 97th percentiles. The dotted horizontal line indicates the lower limit of detection of the assay. Statistical comparison was by t test (P values are shown above the bars). A, Quantities of interleukin (IL)—2, interferon (IFN)—γ, and IL-17. B, Quantities of IL-4, IL-5, and IL- 13. C, Quantities of IL-12, macrophage chemotactic protein (MCP)—1, and IL-6. D, Quantities of IL-1β, IL-7, and IL-10. E, Quantities of granulocyte colonystimulating factor (G-CSF), granulocyte-macrophage (GM)—CSF, and tumor necrosis factor (TNF)—α. F, Quantities of IL-8 and macrophage inflammatory protein (MIP)—1β.
Concentrations of Mediators in Secretions Analyzed by Form and Severity of Illness
We next compared the concentrations of mediators in subjects with URTI versus those with bronchiolitis, for both types of viral infection. No mediator was present in substantially higher concentrations among subjects with LRTI compared with subjects with URTI alone (P ⩾ .04, for each; data not shown). This was the case for subjects with RSV infection as well as for those with influenza virus infection.
We next studied the relationship between mediator release and the measured value of oxygen saturation. The concentrations of the mediators studied bore no substantial relationship with the degree of oxygen saturation (P ⩾ .14, for each).
Persistence of Mediators with Continuing Illness
Study subjects had been wheezing for 1–5 days at the time when samples of secretions were obtained. We wondered whether the concentration of cytokines increased during this interval. Although only 1 sample of secretions was available per subject, we summarized cytokine concentrations by the day after the onset of wheezing on which they had been obtained. The quantities of the T lymphocyte cytokines IL-2, IFN-γ, IL-4, and IL-17 declined over this interval, and most were substantially lower in samples obtained on day 5 than in samples obtained on day 1 (table 2). Therefore, there was no evidence of the development of lymphocyte cytokine responses over this interval.
Table 2.

Concentrations of T lymphocyte cytokines in nasopharyngeal secretions from surviving infants with lower respiratory tract infection grouped by day after onset of wheezing.
Routine Pathological Evaluation of Lung Tissue from Subjects with Fatal LRTI
All autopsy materials underwent routine pathological analysis. Photomicrographs of tissue stained with hematoxylin-eosin (HE) are available in the Appendix. A comparison of pathological findings was done by one pathologist (L.M.), who was unaware of the identity of the infecting agent. In subjects with fatal cases of influenza virus infection, viral antigen was detectable primarily in the epithelium of larger airways (yet severe bronchiolitis was still evident) and, less frequently, in the airway lumen (figure 2A, upper panels). In contrast, in subjects with fatal cases of RSV infection, viral antigen was primarily found in the material obstructing the lumen of the small airways (figure 2B, upper panels). Viral antigen was also present in multinucleated giant cells lining or in the bronchiolar lumen. Viral antigen was much more extensively present in subjects with RSV infection than in those with influenza virus infection. Damage to the bronchiolar epithelium was judged to be much greater in the subjects with RSV infection than in those with influenza virus infection. Periodic acid—Schiff (PAS)—positive material (mucus) was found in the airway in only 1 subjects with influenza virus infection and was absent in the other 19 subjects with RSV and influenza virus infection. Intralumenal material was also negative by Alcian blue staining, indicating the absence of mucins. Other findings were felt to be similar in all 20 subjects, whether or not underlying heart disease or Down syndrome was present. Composites of IHC staining of autopsy tissues from other subjects with fatal cases for viral antigen and all other antigens and enzymes listed below are available in the Appendix.
Figure 2.
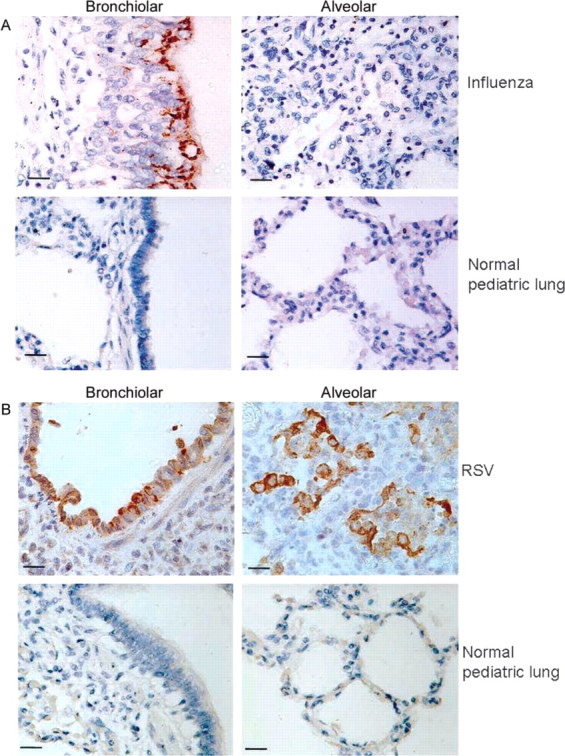
Immunohistochemical (IHC) staining for influenza virus and respiratory syncytial virus (RSV) antigen of bronchiolar and alveolar tissue from infants with lower respiratory tract infection (LRTI) or bronchiolitis and from healthy infants. Autopsy tissues were obtained from human infants with fatal cases of LRTI caused by either influenza virus (A, upper panels) or RSV (B, upper panels). Normal infant lung tissue (from an infant dying of asphyxia) is stained as a control (A and B, lower panels). Brown stain indicates the presence of viral antigen. Influenza virus antigen is found primarily in airway epithelium, whereas RSV antigen is present in both epithelium and in exfoliated epithelial cells plugging the airway. Scale bars show 15 μm; original magnification is ×40.
IHC Analysis
CD4 antigen— and CD8 antigen—positive cells. Surprisingly, CD4 antigen—positive cells were present at a very low frequency in the lungs of subjects with fatal bronchiolitis (or normal infant lung tissue), regardless of the nature of the infecting agent (figure 3, upper panels). CD8 antigen—positive cells also were only rarely detectable in these tissues, again regardless of the nature of the infecting virus (lower panels). CD4 and CD8 cells were readily detectable in tonsil tissue by use of the same IHC techniques. The findings in figure 3 were entirely representative of those in specimens from all 20 subjects with fatal bronchiolitis.
Figure 3.
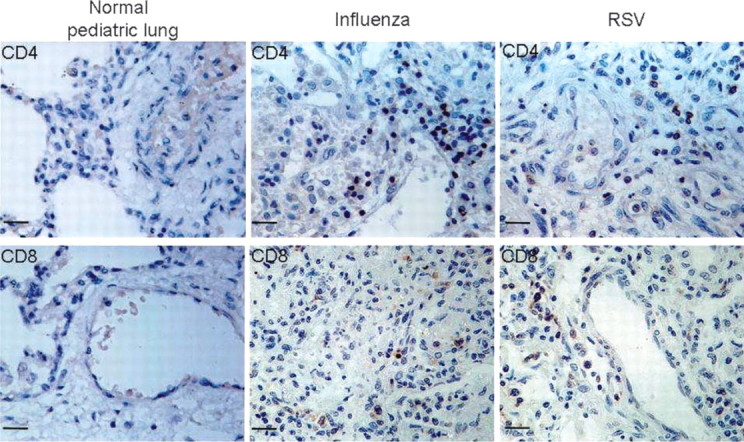
Immunohistochemical staining of lung tissue for CD4- and CD8-positive lymphocytes. Autopsy tissues were stained for cells bearing CD4 (upper panels) or CD8 (lower panels) surface antigens. Normal infant lung tissue is illustrated in the left column. Lung tissue from infants with fatal influenza and respiratory syncytial virus (RSV) infection is displayed in the middle and right columns, respectively. Human tonsilar tissue (not shown) demonstrated numerous cells with positive staining for CD4 and CD8 (positive control).
CD56 antigen—positive cells. The results of IHC staining for CD56 antigen—positive (NK) lymphocytes in the lungs of subjects with fatal LRTI are shown in figure 4. Cells positive for CD56 antigen were rarely detectable in normal infant lungs or in the lungs of subjects with influenza virus or RSV infection. CD56 antigen was readily detectable in stains of a human lung cancer biopsy sample (data not shown).
Figure 4.

Immunohistochemical staining of lung tissue for CD56-positive lymphocytes. Autopsy tissues were studied for CD56 (NK lymphocyte) antigen. Tissues were obtained from an infant dying of asphyxia (normal lung, left panel) and from infants with fatal influenza virus (center panel) or respiratory syncytial virus (RSV) (right panel) infection. Tissue from an adult with lung cancer served as a positive control, demonstrating numerous antigen-positive cells.
Granzyme-positive cells. As is demonstrated in the upper left panel of figure 5, normal infant lung is negative for granzyme, a lymphotoxin. Subjects with influenza virus (upper right) and RSV (lower left) infection were also granzyme negative. The expected positive staining for granzyme of lymphocytes present in a biopsy sample from a lung cancer patient is demonstrated in the lower right panel.
Figure 5.
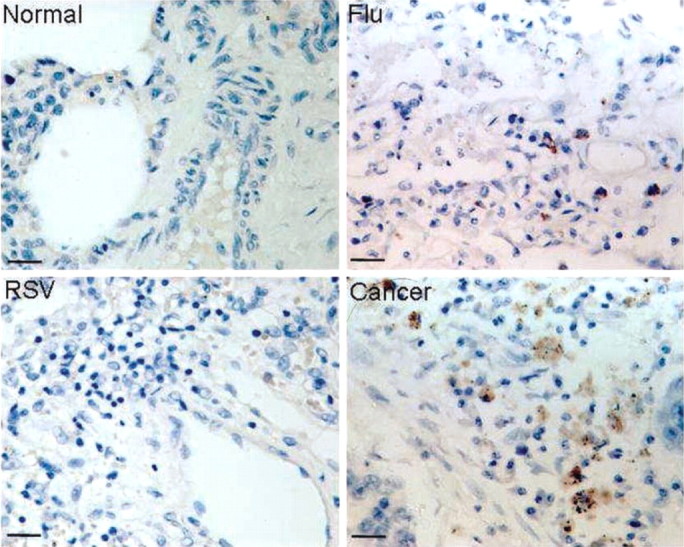
Immunohistochemical staining of lung tissue for granzyme. Lung tissues were stained for the presence of granzyme, a lymphocyte product that mediates cytotoxic reactions against infected and stressed cells. Tissue was obtained from an infant dying of asphyxia (normal lung, upper left panel). An infant with fatal influenza virus infection is represented in the upper right panel. An infant with fatal respiratory syncytial virus (RSV) infection is represented in the lower left panel, and an adult with lung cancer is represented in the lower right panel.
CD16 antigen—positive cells. Lung tissues from infants with fatal RSV and influenza virus LRTI were studied for expression of the neutrophil and macrophage antigen CD16 (figure 6). CD16 antigen was not observed in normal infant lung tissue (left panel). In contrast, strongly positive staining was observed in subjects with RSV or influenza virus infection (center and right panels). Staining was noted in inflammatory cells in the alveolar space and interstitium and in cells within the exudate that obstructed the bronchiolar lumen.
Figure 6.

Immunohistochemical staining of lung tissue for CD16. Lung tissues were studied for CD16 (expressed primarily on granulocytes and macrophages) antigen. Tissues were obtained from an infant dying of asphyxia (normal lung, left panel) and from infants with fatal influenza virus (center panel) or respiratory syncytial virus (RSV; right panel) infection.
Caspase 3 staining. We next stained lung tissues for the apoptosis marker caspase 3 (figure 7). Normal infant lung tissue (left panel) did not demonstrate positive staining for caspase 3. Lung tissue from infants with fatal LRTI showed strong staining. For fatal RSV infection, caspsase 3 staining was most evident in bronchiolar epithelial cells (right panel). For influenza cases, staining was most prominent in inflammatory cells (macrophages and neutrophils, center panel).
Figure 7.

Immunohistochemical staining of lung tissue for caspase 3. Lung tissues were stained for the presence of the apoptosis marker caspase 3. Tissue from an infant dying of asphyxia (normal lung, left panel) served as a negative control. Staining in cases of influenza virus infection was primarily observed in inflammatory cells (center panel). Subjects with respiratory syncytial virus (RSV) infection demonstrated positive staining in bronchiolar epithelial cells (right panel).
Discussion
Among infants surviving viral LRTI, most of the 17 cytokines measured were present in lower quantities in subjects with RSV infection than in those with influenza virus infection, and none were present at higher concentrations in those with RSV infection. Specifically, there was no evidence of T lymphocyte activation in the respiratory secretions from these infants at the time of hospitalization for RSV LRTI; that is, the classical T lymphocyte cytokines IL-2, IL-4, IFN-γ, and IL-17 were nearly undetectable in the secretions (figure 1A and 1B). In contrast, IL-2, IL-4, IFN-γ, and IL-17 were all detected more frequently or at higher concentrations in subjects with influenza virus LRTI. Because infants with RSV infection had more-severe illness (reduced oxygen saturation and more-prolonged hospital stays) than did infants with influenza virus bronchiolitis, these findings therefore challenge the idea that overresponsiveness of cytotoxic lymphocytes plays a critical role in the development of bronchiolitis [3–11]. All samples of secretions for cytokine analysis were obtained by the same personnel using the same techniques during the same winter season. Samples were processed and analyzed in an identical fashion. Subjects with RSV infection were similar in age to those with influenza virus infection and had apparently been ill for 1 more day, on average, than had subjects with influenza virus infection (table 1). Therefore, these factors would not seem to account for the reduced cytokine responses observed in subjects with RSV infection.
The quantities of most other cytokines and chemokines were also reduced for RSV infection in comparison to influenza virus infection, perhaps because their synthesis was not being stimulated in the absence of a T lymphocyte response. A few cytokines were present in equal concentrations in subjects with RSV or influenza virus infection. Possible alternative sources of these mediators include the respiratory epithelium and other inflammatory cells. Previous studies of cytokine responses in infantile bronchiolitis have found lower concentrations of IFN-γ in subjects with more-severe forms of illness [27–29], which is generally consistent with the findings of our study. The present study additionally demonstrated the limited presence of the lymphocyte cytokines IL-2 and IL-17 in bronchiolitis.
In addition to the low quantities of lymphocyte cytokines in secretions from surviving subjects, lymphocytes expressingCD4 or CD8 antigen were present only in very low numbers in lung tissue. Granzyme-positive cytotoxic lymphocytes also were not detectable in the lungs of infants with fatal LRTI. In previous studies of bronchoalveolar lavage (BAL) fluid recovered from infants with bronchiolitis, <2% of cells present in BAL fluids were positive for CD8 antigen [21, 30]. In the present study, lymphocytes bearing CD4, CD8, or CD56 antigen were rarely observed in lung autopsy tissues. In contrast, in subjects surviving measles virus pneumonia, CD8 antigen—positive lymphocytes predominate in BAL fluids [31]. In fatal influenza virus infections beyond infancy, lymphocytes predominate in lung tissue [32, 33] and are also present in infections with severe acute respiratory syndrome coronavirus [34]. Although the characterization of the mononuclear cell infiltrate observed in lung tissue from subjects with fatal adenovirus infections is limited, nevertheless, CD4- and CD8-bearing lymphocytes proliferate in peripheral blood and release IFN-γ [35, 36]. Thus, the lack of a CD8 lymphocyte response in RSV and influenza virus infections in infants appears to be aberrant. We are unaware of studies evaluating granzyme staining in immunologically normal infants who present with LRTI caused by other viruses.
These overall findings strongly suggest that T lymphocyte activation and cytotoxic activity do not occur in the lungs at the time when infants with bronchiolitis are experiencing their most-severe symptoms. Low numbers of cells expressing CD56 antigen were detected, suggesting that NK cell activity was also diminished. Whether adaptive responses would have developed later in subjects who died (or survived) is moot, but these responses were not present at the time of maximum illness.
Studies of the development of RSV-specific cytotoxic activity in peripheral blood lymphocytes after RSV infection have been able to demonstrate only weak responses [37, 38]. Interestingly, several reports have described mechanisms by which RSV infection can inhibit the development of cytotoxic responses in vivo, including interference with antigen-dependent T cell receptor signaling [39], alteration of dendritic cell function [40], induction of lymphocyte apoptosis [41], and suppression of CD4 lymphocyte function by IFN-α and IFN-λ [42].
Expression of RSV antigen (detected by IHC analysis) was judged to exceed that of influenza virus antigen in subjects with fatal cases of bronchiolitis caused by the respective virus. Although quantitative morphometry was not available, these observations were made by a pathologist reviewing slides without knowledge of the nature of the infecting virus. Previous studies of RSV bronchiolitis have found that higher viral loads (assessed by quantitative polymerase chain reaction) were associated with more-severe disease [43].
Finally, staining for the apoptosis marker caspase 3 was strong in subjects with fatal cases of bronchiolitis. However, for RSV infection, caspase 3 and viral antigen were detected in bronchiolar epithelial cells. This suggests that activation of apoptotic mechanisms may be critical to clearing RSV-infected epithelial cells when cytotoxic T cell responses are absent. In contrast, caspase 3 staining was more prominent in macrophages and neutrophils in subjects with fatal cases of influenza virus infection. Although definitive conclusions are not possible, one possible explanation of the findings is that the expression of viral antigen was already declining in subjects of influenza virus infection (and bronchiolar epithelium was more intact) and that apoptosis was being induced in macrophages and neutrophils to reduce the inflammatory response. Further study of this critical area is needed. Other models of lung injury demonstrate that apoptotic signals are expressed on damaged epithelium immediately after toxic injury to the lungs. However, after the toxin has been cleared, apoptosis is prominent in inflammatory cells, presumably reflecting an attempt to terminate inflammation [44].
Limitations of the present study include the fact that cytokines were measured only once during the time of acute illness. We attempted to correct for this deficiency by studying the kinetics of mediator responses in samples grouped by day 1–5 of illness. Quantities of IFN-γ, IL-4, and IL-17 declined over this interval. Although this suggests that we did not miss the eventual development of lymphocyte cytokine responses, follow-up samples obtained over several days would have been preferable. Another limitation is that samples of secretions for cytokine analysis, viral quantitation, and pathological analysis were not available from the same subjects. Nevertheless, bronchiolitis as a disease entity is thought to be similar in presentation worldwide [45–47], so regional differences in pathogenesis are unlikely. More importantly, the findings from the 2 populations in the present study are consistent in suggesting that T lymphocyte responses are reduced or absent in moresevere forms of infantile viral LRTI that result in hypoxia, the need for hospitalization, or death.
In a previous study [25], quantities of 6 cytokines (IL-12, TNF-α, IL-6, IL-1β, IL-8, and IL-10) were found to be similar in infants with RSV infection and those with influenza virus infection. However, quantities of cytokines were far lower in that study than in the present study, perhaps because the samples were nasal washes and not aspirates. Other reports have found lower quantities of TNF-α [48], MCP-1, and MIP-1α [49] in infants with RSV infection than in those with influenza virus infection.
In summary, we propose that the pathogenesis of viral LRTI is primarily related to the failure to develop an adaptive cytotoxic T lymphocyte response. Instead, clearance of the virus apparently depends on inefficient innate immune responses mediated by macrophages and neutrophils. The expression of caspase 3 in the lungs suggests that apoptotic mechanisms are activated in an attempt to clear virus. These mechanisms may eventually lead to the eradication of infection, but at the expense of damage to uninfected neighboring epithelial cells. These findings should encourage the testing of antiviral compounds with activity against RSV and influenza virus in infantile LRTI.
Footnotes
Potential conflicts of interest: none reported.
Presented in part: American Thoracic Society International Conference, San Diego, 20–24 May 2006; Pediatric Academic Societies Annual Meeting, San Francisco, 3–6 May 2006.
Financial support: National Institute of Allergy and Infectious Diseases (grant P01 AI 062885 to R.P.G.); National Heart, Lung, and Blood Institute (grant N01 HV28184 to R.P.G.).
References
- 1.Leader S, Kohlhase K. Respiratory syncytial virus-coded pediatric hospitalizations, 1997 to 1999. Pediatr Infect Dis J. 2002;21:629–32. doi: 10.1097/00006454-200207000-00005. [DOI] [PubMed] [Google Scholar]
- 2.Garenne M, Ronsmans C, Campbell H. The magnitude of mortality from acute respiratory infections in children under 5 years in developing countries. World Health Stat Q. 1992;45:180–91. [PubMed] [Google Scholar]
- 3.Openshaw PJM, Tregoning JS. Immune responses and disease enhancement during respiratory syncytial virus infection. Clin Microbiol Rev. 2005;18:541–55. doi: 10.1128/CMR.18.3.541-555.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graham BS, Rutigliano JA, Johnson TR. Respiratory syncytial virus immunobiology and pathogenesis. Virology. 2002;297:1–7. doi: 10.1006/viro.2002.1431. [DOI] [PubMed] [Google Scholar]
- 5.McNamara PS, Smyth R. The pathogenesis of respiratory syncytial virus disease in childhood. Brit Med Bull. 2002;61:13–28. doi: 10.1093/bmb/61.1.13. [DOI] [PubMed] [Google Scholar]
- 6.Varga SM, Braciale TJ. RSV-induced immunopathology: dynamic interplay between the virus and host immune response. Virology. 2002;295:203–7. doi: 10.1006/viro.2002.1382. [DOI] [PubMed] [Google Scholar]
- 7.Durbin JE, Durbin RK. Respiratory syncytial virus-induced immunoprotection and immunopathology. Viral Immunol. 2004;17:370–80. doi: 10.1089/vim.2004.17.370. [DOI] [PubMed] [Google Scholar]
- 8.Tripp RA. Pathogenesis of respiratory syncytial virus infection. Viral Immunol. 2004;17:165–81. doi: 10.1089/0882824041310513. [DOI] [PubMed] [Google Scholar]
- 9.Graham BS, Bunton LA, Wright PF, Karzon DT. Role of T lymphocyte subsets in the pathogenesis of primary infection and rechallenge with respiratory syncytial virus in mice. J Clin Invest. 1991;88:1026–33. doi: 10.1172/JCI115362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ostler T, Davidson W, Ehl S. Virus clearance and immunopathology by CD8+ T cells during infection with respiratory syncytial virus are mediated by IFNγ. Eur J Immunol. 2002;32:2117–23. doi: 10.1002/1521-4141(200208)32:8<2117::AID-IMMU2117>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 11.Pinto RA, Arredondo SM, Bono MR, Gaggero AA, Diaz PV. T helper 1/T helper 2 cytokine imbalance in respiratory syncytial virus infection is associated with increased endogenous plasma cortisol. Pediatrics. 2006;117:e878–86. doi: 10.1542/peds.2005-2119. [DOI] [PubMed] [Google Scholar]
- 12.Kim HW, Canchola JG, Brandt CD, et al. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am J Epidemiol. 1969;89:422–34. doi: 10.1093/oxfordjournals.aje.a120955. [DOI] [PubMed] [Google Scholar]
- 13.Kim HW, Leikin SL, Arrobio J, Brandt CD, Chanock RM, Parrott RH. Cell-mediated immunity to respiratory syncytial virus induced by inactivated vaccine or by infection. Pediatr Res. 1976;10:75–8. doi: 10.1203/00006450-197601000-00015. [DOI] [PubMed] [Google Scholar]
- 14.Neilson KA, Yunis EJ. Demonstration of respiratory syncytial virus in an autopsy series. Pediatr Pathol. 1990;10:491–502. doi: 10.3109/15513819009067138. [DOI] [PubMed] [Google Scholar]
- 15.Adams JM, Imagawa DT, Zike K. Epidemic bronchiolitis and pneumonitis related to respiratory syncytial virus. JAMA. 1961;176:1037–9. doi: 10.1001/jama.1961.63040250020020b. [DOI] [PubMed] [Google Scholar]
- 16.Whimbey E, Couch RB, Englund JA, et al. Respiratory syncytisal virus pneumonia in hospitalized adult patients with pneumonia. Clin Infect Dis. 1995;21:376–9. doi: 10.1093/clinids/21.2.376. [DOI] [PubMed] [Google Scholar]
- 17.Whimbey E, Champlin RE, Couch RB, et al. Community respiratory virus infections among hospitalized adult bone marrow transplant recipients. Clin Infect Dis. 1996;22:778–82. doi: 10.1093/clinids/22.5.778. [DOI] [PubMed] [Google Scholar]
- 18.Wohl MEB, Chernick V. Bronchiolitis. Am Rev Respir Dis. 1978;118:759–81. doi: 10.1164/arrd.1978.118.4.759. [DOI] [PubMed] [Google Scholar]
- 19.Hall CB. Respiratory syncytial virus and parainfluenza virus. N Engl J Med. 2001;344:1917–28. doi: 10.1056/NEJM200106213442507. [DOI] [PubMed] [Google Scholar]
- 20.Hall CB, Hall WJ, Spears DM. Clinical and physiological manifestations of bronchiolitis and pneumonia: outcome of respiratory syncytial virus. Am J Dis Child. 1979;133:798–802. doi: 10.1001/archpedi.1979.02130080038006. [DOI] [PubMed] [Google Scholar]
- 21.Everard ML, Swarbrick A, Wrightham M, et al. Analysis of cells obtained by bronchial lavage of infants with respiratory syncytial virus infection. Arch Dis Child. 1994;71:428–32. doi: 10.1136/adc.71.5.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim CK, Chung CY, Choi SJ, Kim DK, Park X, Koh YY. Bronchoalveolar lavage cellular composition in acute asthma and acute bronchiolitis. J Pediatr. 2000;137:517–22. doi: 10.1067/mpd.2000.108392. [DOI] [PubMed] [Google Scholar]
- 23.Graham BS, Perkins MD, Wright PF, Karzon DT. Primary respiratory syncytial virus infection in mice. J Med Virol. 1988;26:153–62. doi: 10.1002/jmv.1890260207. [DOI] [PubMed] [Google Scholar]
- 24.Wright PF, Gruber WC, Peters M, et al. Illness severity, viral shedding, and antibody responses in infants hospitalized with bronchiolitis caused by respiratory syncytial virus. J Infect Dis. 2002;185:1011–8. doi: 10.1086/339822. [DOI] [PubMed] [Google Scholar]
- 25.Laham FR, Israele V, Casellas JM, et al. Differential production of inflammatory cytokines in primary infection with human metapneumovirus and with other common respiratory viruses of infancy. J Infect Dis. 2004;189:2047–56. doi: 10.1086/383350. [DOI] [PubMed] [Google Scholar]
- 26.Wolf DG, Greenberg D, Kalkstein D, et al. Comparison of human metapneumovirus, respiratory syncytial virus and influenza A virus lower respiratory tract infections in hospitalized young children. Pediatr Infect Dis J. 2006;25:320–4. doi: 10.1097/01.inf.0000207395.80657.cf. [DOI] [PubMed] [Google Scholar]
- 27.Garofalo RP, Patti J, Hintz KH, Hil V, Ogra PL, Welliver RC. Macrophage inflammatory protein—1α (not T helper type 2 cytokines) is associated with severe forms of respiratory syncytial virus bronchiolitis. J Infect Dis. 2001;184:393–9. doi: 10.1086/322788. [DOI] [PubMed] [Google Scholar]
- 28.Bont L, Heijnen CJ, Kavelaars A, et al. Local interferon-γ levels during respiratory syncytial virus lower respiratory tract infection are associated with disease severity. J Infect Dis. 2001;184:355–8. doi: 10.1086/322035. [DOI] [PubMed] [Google Scholar]
- 29.Aberle JH, Aberle SW, Dworzak MN, et al. Reduced interferon-γ expression in peripheral blood mononuclear cells of infants with severe respiratory syncytial virus disease. Am J Respir Crit Care Med. 1999;160:1263–8. doi: 10.1164/ajrccm.160.4.9812025. [DOI] [PubMed] [Google Scholar]
- 30.Smith PK, Wang S-Z, Dowling KD, Forsyth KD. Leucocyte populations in respiratory syncytial virus-induced bronchiolitis. J Paediatr Child Health. 2001;37:146–51. doi: 10.1046/j.1440-1754.2001.00618.x. [DOI] [PubMed] [Google Scholar]
- 31.Myou S, Fujimara M, Ueno T, Matsuda T. Bronchoalveolar lavage cell analysis in measles viral pneumonia. Eur Respir J. 1993;6:1437–42. [PubMed] [Google Scholar]
- 32.Mulder J, Hers JFP. Leiden, The Netherlands: Walters-Noordhoff, International School Book Service; 1972. Influenza. [Google Scholar]
- 33.Writing Committee of the World Health Organization Consultation on Human Influenza A/H5. Current concepts: avian influenza (H5N1) infection in humans. N Engl J Med. 2005;353:1374–85. doi: 10.1056/NEJMra052211. [DOI] [PubMed] [Google Scholar]
- 34.Lee N, Hui D, Wu A, et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N Engl J Med. 2003;348:1986–94. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 35.Flomenberg P, Piaskowski V, Truitt RL, Casper JT. Characterization of human proliferative T cell responses to adenovirus. J Infect Dis. 1995;171:1090–6. doi: 10.1093/infdis/171.5.1090. [DOI] [PubMed] [Google Scholar]
- 36.Matsubara T, Inoue T, Tashiro N, Katayama K, Matsuoka T, Furukawa S. Activation of peripheral blood CD8+ T cells in adenovirus infection. Pediatr Infect Dis J. 2000;19:766–8. doi: 10.1097/00006454-200008000-00023. [DOI] [PubMed] [Google Scholar]
- 37.Mbawuike INM, Wells J, Byrd R, Cron SG, Glezen WP, Piedra PA. HLA-restricted CD8+ cytotoxic T lymphocyte, interferon-γ, and interleukin-4 responses to respiratory syncytial virus infection in infants and children. J Infect Dis. 2001;183:687–96. doi: 10.1086/318815. [DOI] [PubMed] [Google Scholar]
- 38.Isaacs D, Bangham CR, McMichael AJ. Cell-mediated cytotoxic response to respiratory syncytial virus in infants with bronchiolitis. Lancet. 1987;2:769–71. doi: 10.1016/s0140-6736(87)92502-5. [DOI] [PubMed] [Google Scholar]
- 39.Chang J, Braciale TJ. Respiratory syncytial virus infection suppresses lung CD8+ T-cell effector activity and peripheral CD8+ T-cell memory in the respiratory tract. Nature Med. 2002;8:54–60. doi: 10.1038/nm0102-54. [DOI] [PubMed] [Google Scholar]
- 40.Guerrero-Plata A, Casola A, Suarez G, et al. Differential response of dendritic cells to human metapneumovirus and respiratory syncytial virus. Am J Respir Cell Mol Biol. 2006;34:320–9. doi: 10.1165/rcmb.2005-0287OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roe JFE, Bloxham DM, White DK, Ross-Russell RI, Tasker RTC, OȧDonnell DR. Lymphocyte apoptosis in acute respiratory syncytial virus bronchiolitis. Clin Exp Immunol. 2004;137:139–45. doi: 10.1111/j.1365-2249.2004.02512.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chi B, Dickensheets HL, Spann KM, et al. Alpha and lambda interferon together mediate suppression of CD4 T cells induced by respiratory syncytial virus. J Virol. 2006;80:5032–40. doi: 10.1128/JVI.80.10.5032-5040.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeVincenzo JP, El Saleeby CM, Bush AJ. Respiratory syncytial virus load predicts disease severity in previously healthy infants. J Infect Dis. 2005;191:1861–8. doi: 10.1086/430008. [DOI] [PubMed] [Google Scholar]
- 44.Hussain N, Wu F, Zhu L, Thrall R, Kresch MJ. Neutrophil apoptosis during the development and resolution of loeic acid-induced acute lung injury in the rat. Am J Respir Cell Mol Biol. 1998;19:867–74. doi: 10.1165/ajrcmb.19.6.3118. [DOI] [PubMed] [Google Scholar]
- 45.Chan PWK, Chew FT, Tan TN, Chua KB, Hooi PS. Seasonal variation in respiratory syncytial virus chest infection in the tropics. Pediatr Pulmonol. 2002;34:47–51. doi: 10.1002/ppul.10095. [DOI] [PubMed] [Google Scholar]
- 46.Weber MW, Dackour R, Usen S, et al. The clinical spectrum of respiratory syncytial virus disease in The Gambia. Pediatr Infect Dis J. 1998;17:224. doi: 10.1097/00006454-199803000-00010. [DOI] [PubMed] [Google Scholar]
- 47.Orstavik I, Carlsen K-H, Halvorsen K. Respiratory syncytial virus infections in Oslo 1972–1978. Acta Paediatr Scand. 1980;69:717–22. doi: 10.1111/j.1651-2227.1980.tb07139.x. [DOI] [PubMed] [Google Scholar]
- 48.Sung RTS, Hui SHL, Wong CW, Lam CWK, Yin J. A comparison of cytokine responses in respiratory syncytial virus and influenza infections in infants. Eur J Pediatr. 2001;160:117–22. doi: 10.1007/s004310000676. [DOI] [PubMed] [Google Scholar]
- 49.Garofalo RP, Hintz KH, Hill V, Patti J, Ogra PL, Welliver RC., Sr A comparison of epidemiologic and immunologic features of bronchiolitis caused by influenza virus and respiratory syncytial virus. J Med Virol. 2005;75:282–9. doi: 10.1002/jmv.20268. [DOI] [PubMed] [Google Scholar]