Interaction modules that impart specificity to disordered protein
. Author manuscript; available in PMC: 2024 May 1.
Published in final edited form as: Trends Biochem Sci. 2023 Feb 6;48(5):477–490. doi: 10.1016/j.tibs.2023.01.004
Abstract
Intrinsically disordered regions (IDRs) are especially enriched among proteins that regulate chromatin and transcription. As a result, mechanisms that influence specificity of IDR-driven interactions have emerged as exciting unresolved issues for understanding gene regulation. Here we review the molecular elements frequently found within IDRs that confer regulatory specificity. In particular, we review the differing roles of disordered low-complexity regions (LCRs) and short linear motifs (SLiMs) towards selective nuclear regulation. Examination of IDR-driven interactions highlights SLiMs as organizers of selectivity, with widespread roles in gene regulation and integration of cellular signals. Analysis of recurrent interactions between SLiMs and folded domains suggests diverse avenues for SLiMs to influence phase-separated condensates and highlights opportunities to manipulate these interactions for control of biological activity.
Keywords: LLPS, structure, condensate, composition, expression
The origins of selectivity in IDR-driven processes
Interest in the properties and functional roles of intrinsically disordered regions (IDRs; see Glossary) has grown considerably over the past decade. Modern structural biology approaches [1,2] and computational tools [3–6] have revealed that many proteins possess a high proportion of IDRs rather than stably folded domains. Indeed, 37–50% of the human proteome consists of disordered sequences [3]. However, due to several challenges specific to flexible and disordered regions (Box 1), the functional characterization of unstructured protein regions has lagged, and IDRs are thus arguably the most under-characterized portion of the eukaryotic proteome.
Box 1. Opportunities to improve the study of intrinsically disordered regions (IDRs).
IDRs contribute to the dynamic organization of protein networks inside cells. Although characterization of such protein networks using conventional methods has remained challenging, each of the following major challenges represents an opportunity to transform research in this area: (1) interactions between factors are often short-lived [149], and traditional biochemical approaches (e.g. co-immunoprecipitation, genetic manipulation) have limited temporal resolution, which obscures detection of these fast activities; (2) recapitulating short-lived interaction networks outside of the native cellular environment remains difficult and results are subject to interpretation; (3) eukaryotic proteins containing disordered domains are often unstable in vitro and can be difficult to purify due to aggregation and sensitivity to proteolytic cleavage [150]; (4) even when successfully purified, IDRs and the conformationally heterogenous interactions mediated by them often cannot be resolved to high resolution using cryoEM and X-ray crystallography; and (5) computational identification of selective interaction motifs like SLiMs based on sequence similarity is generally subject to statistical uncertainty and requires experimental validation.
In spite of these challenges, new methods have proven to be powerful tools for IDR characterization. Such approaches include the use of high-throughput peptide arrays [151], or proteomic peptide phage display libraries that enable large-scale assessment of interactions of proteins of interest with potential SLiMs in the human proteome [152]. Additionally, motif discovery tools capable of scanning for degenerate consensus motifs proteome-wide [153] can be used to tease out these motifs based on putative patterns, which can also be predicted de novo using computational approaches [154–156]. The wide range of interdisciplinary approaches in this area suggests that new technologies will play important roles in overcoming the above challenges.
Despite lacking stable three-dimensional structure, IDRs have emerged as functionally important elements that complement folded protein domains for control of biological processes [7,8]. IDRs harbor interfaces for selective interactions with folded domains and, in some cases, engage other proteins through multivalent interactions that facilitate cooperative demixing via phase separation. While diverse phase separation phenomena influence cellular function, recent findings have underscored the pervasive role of nuclear compartmentalization via liquid-liquid phase separation (LLPS, reviewed in [9–11]). However, many questions remain about the interplay between soluble factors and those in phase separated condensates, as well as the ability of IDRs to regulate distinct cellular processes with selectivity.
Here we focus on the roles of IDRs in chromatin and transcription regulators, which are highly enriched with disordered regions. We highlight aspects of chromatin and transcription where IDRs influence selectivity, review recurrent functional elements found in IDRs of transcription regulators, and outline the key settings where these functional elements mediate specific interactions. Finally, we address how interactions involving IDRs can be harnessed for therapeutic targeting.
IDRs are ubiquitous and mediate specific nuclear processes
Chromatin and transcription regulators are enriched in disorder
The proportion of folded structure within human proteins bears a relationship to biological function [12]. Proteins abundant in IDRs are most common among factors that regulate chromatin or transcription (Figure 1A). In contrast, proteins composed mostly of folded domains are enriched among metabolic enzymes and proteins regulating homeostasis. Protein disorder is thus uniquely enriched in chromatin and transcription regulators among all cellular processes.
Figure 1. Intrinsically disordered regions (IDRs) are enriched among chromatin and transcription regulators.
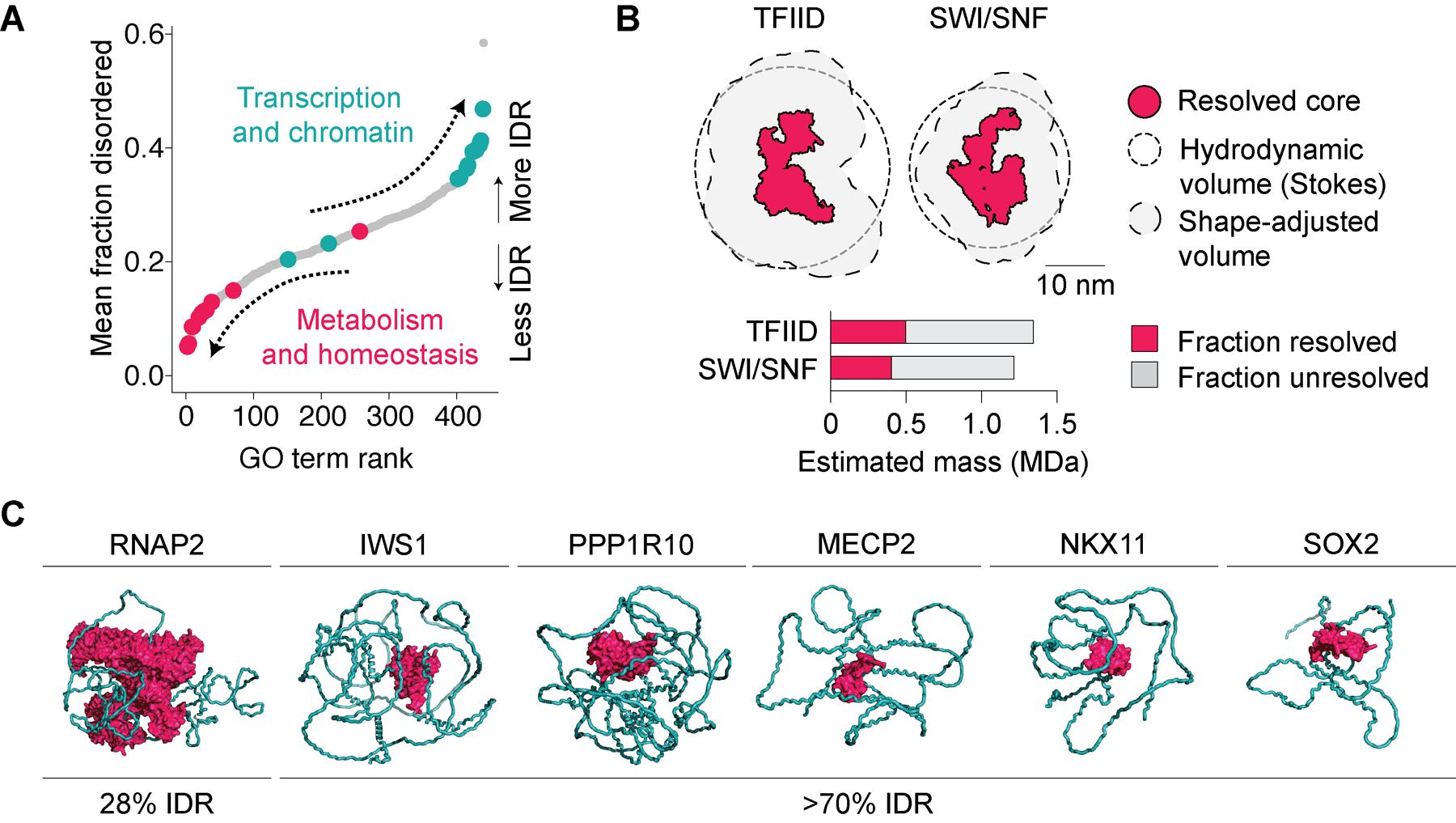
(A) All UniProt GO terms containing ≥50 human proteins, ranked by mean percent disorder in proteins contained within each term. GO terms related to transcription and chromatin are highlighted in teal and those related to regulation of metabolism and homeostasis in fuchsia. (B) Illustration of resolved (fuchsia) and unresolved (missing) regions in multiprotein complexes resolved by Cryo-EM (PDB accessions 6MZL and 6LTJ). Stokes hydrodynamic volumes of protein are based on molecular weight and sedimentation constants of full complexes, illustrating the large portion of volume made up by unresolved regions. (C) Example full-length structures of chromatin and transcription regulators predicted by AlphaFold2. Disordered regions in depicted structures are artificially constrained into a more compact configuration in AlphaFold2 for illustration purposes. Folded domains are highlighted in fuchsia and IDRs in teal.
New approaches in structure determination have revealed the ordered core regions of many complexes critical to transcription regulation [13–18] as well as higher-order assemblies [16,19,20]. The structures of these core complexes provide deep insights into the physical basis for the activities of the transcription machinery, but also depict a fraction of the full mass of each complex (Figure 1B). Because intrinsically disordered and dynamic regions remain largely hidden from view, many questions remain regarding how unstructured sequences modulate the activities of these and other transcriptional regulators.
Among transcriptional regulators, the abundance of disorder varies considerably. Many human transcription regulators are predicted to be composed of >70% IDRs (Figure 1C) [4,5]. Examples of such proteins include Mediator subunit MED9, transcription elongation regulators IWS1 and PPP1R10, splicing factor SRSF2, transcriptional repressor MECP2, or transcription factors (TFs) SOX2 and JUND (Figure 1C) [4]. The high fraction of IDRs specific to chromatin and transcriptional regulators has remained a vexing question for many years [7], suggesting that a full understanding of transcription will require untangling the functional roles played by IDRs.
IDRs are functional and influence selectivity of gene regulation
Despite the lack of stable structure, many IDRs are able to provide selectivity. As described in greater detail below, this selectivity arises from a spectrum of interactions that span both soluble protein-protein interactions and those involving LLPS. The roles of IDRs in transcription regulation are generally related to one of four non-mutually exclusive activities: (1) regulating subnuclear localization; (2) organizing rapidly switchable protein-protein interactions that regulate protein activity; (3) acting as degrons or mediating degron masking to regulate protein stability; and/or (4) assisting assembly of oligomeric and higher-order complexes (Figure 2).
Figure 2. Principal mechanisms by which IDRs selectively contribute to transcription regulation.
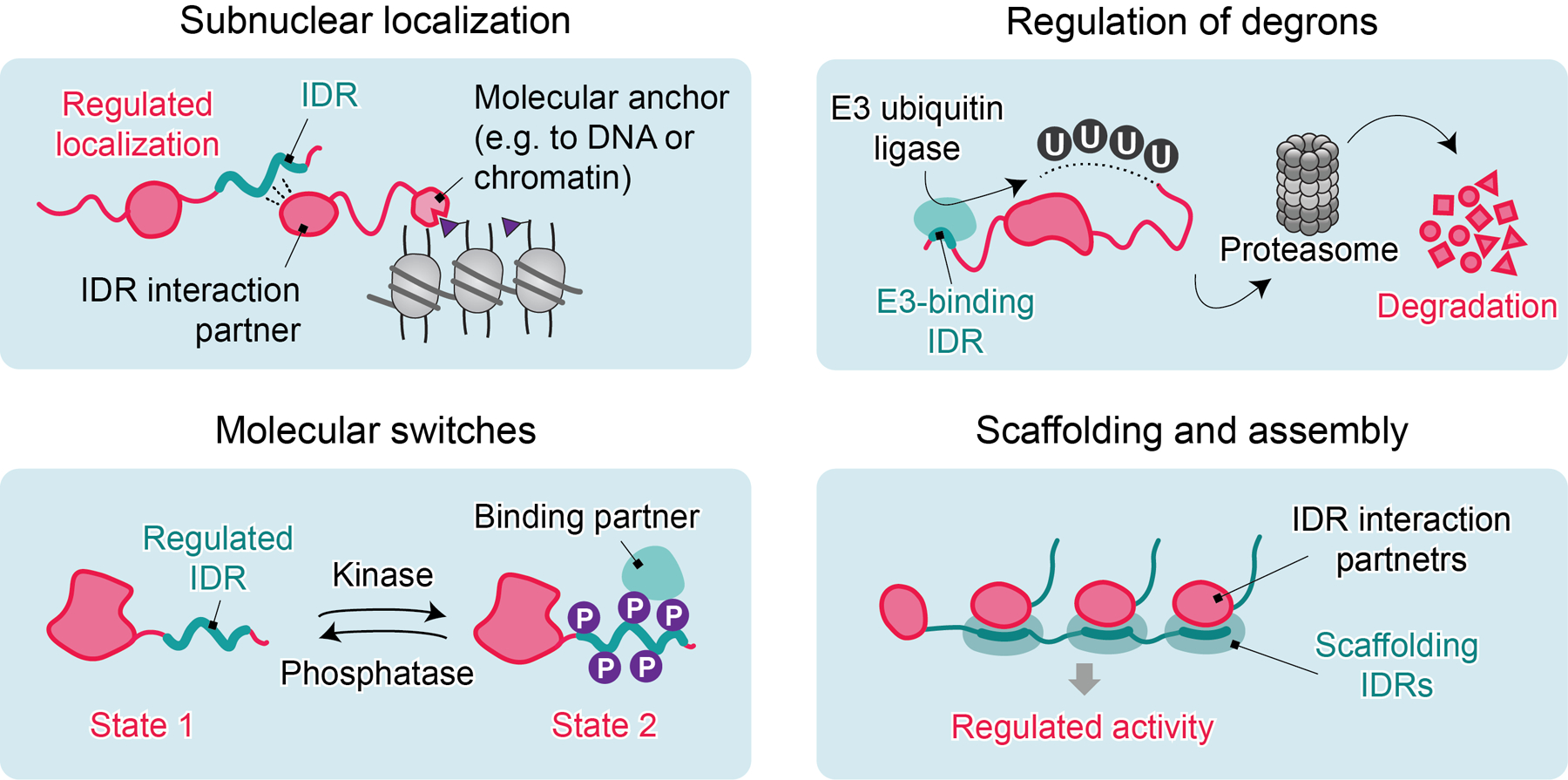
Selective roles of IDRs are generally related to the non-mutually exclusive activities that include: regulation of subnuclear localization; organization of switchable protein-protein interactions to influence other proteins or to auto-regulate; acting as degrons or mediating degron masking to regulate protein stability; and/or assisting assembly of oligomeric and higher-order complexes.
Subnuclear localization
A major avenue by which IDRs influence subnuclear localization is through selective partitioning of proteins into diverse phase-separated condensates (reviewed in [9,21,22]). Partitioning of proteins via LLPS can produce >10,000-fold increases in their local concentrations in the absence of a membrane [23], and such condensates exhibit the hallmark ability to undergo fission and fusion [24]. Additionally, the concentrations of individual components containing IDRs in multicomponent phase-separating systems influence condensate composition [25]. Hence LLPS confers distinct physicochemical properties beyond conventional protein-protein interactions.
IDRs from many chromatin regulators are sufficient to induce LLPS in vitro and in vivo, including a large number of TFs [26–28], BRD4 [29], Mediator [29,30], splicing and elongation factors [31,32], RNAP2 [33,34] and others. The selectivity of LLPS is exemplified by the existence of two types of transcription-associated condensates with distinct composition, referred to as “initiation condensates” (also known as “Mediator condensates” or “super-enhancer condensates”) [22,26,35] and “elongation/RNA processing condensates” [31,32,36]. However, the molecular basis for the selectivity of transcriptional condensates remains an area of active investigation.
One of the original chromatin features posited to undergo LLPS is heterochromatin, which is facilitated by the heterochromatin protein 1 (HP1) protein family that reads trimethylated histone H3 on lysine 9 (H3K9me3). These interactions influence gene silencing in part by excluding access to the transcription machinery [37–39]. Unlike HP1β and HP1γ [40], HP1α is capable of LLPS upon N-terminal phosphorylation or in the presence of DNA in vitro [40,41], and it has been suggested that HP1 promotes heterochromatin partitioning through LLPS [41–43]. However, several points challenge an LLPS-driven mechanism of heterochromatin gene silencing. For example, compaction of heterochromatin foci is independent of HP1 in living cells [44]. Moreover, genetic knockout of the HP1α isoform most prone to LLPS does not lead to global chromatin decompaction [45], and biallelic HP1α knockout mice are viable and fertile [46]. Finally, unlike other IDR-containing proteins [47], HP1 has only a weak capacity to form artificially induced LLPS droplets in living cells [44]. These observations raise questions regarding the necessity or sufficiency of LLPS as a driving mechanism underlying heterochromatin dynamics.
IDRs can also influence subnuclear localization of transcription regulators through conventional bipartite interactions (i.e. those that occur between soluble proteins without LLPS). Such interactions are generally mediated by selective binding of IDRs to well-folded domains and play unambiguous functional roles in gene regulation. Prominent examples of such IDRs include histone tails, nuclear localization signals [48], disordered transactivation domains in TFs that selectively recruit co-factors [49], or unstructured motifs in chromatin scaffolds [50,51]. Several examples of ordered and disordered protein elements that mediate these specific interactions are discussed in later sections.
Molecular switches
Many IDRs maintain selective interactions regulated by post-transitional modifications (PTMs) and hence possess switch-like properties (Figure 2). Signal-dependent IDRs can promote exchange of proteins between distinct LLPS condensates. For example, serine-2 phosphorylation of RNAP2 C-terminal domain (CTD) mediates the exchange of RNAP2 from initiation condensates to elongation condensates with distinct protein composition [31,32]. Similarly, condensation of a BRD4 isoform is reduced by its phosphorylation [52]. These examples of signal-dependent plasticity illustrate the capacity of IDR-driven LLPS to organize related nuclear activities and implicate PTMs as important contributors to selective associations with distinct condensate types.
PTM-regulated IDR switches are also capable of promoting specific nuclear interactions using conventional protein-protein interactions where LLPS is not a strict functional requirement. Examples of such IDRs include those found in KMT2A, JPO2, POGZ, MED1, and IWS1, which all compete for the same interaction domain on the H3K36me3 reader LEDGF (PSIP1) [53–55]. These interactions are regulated by IDR phosphorylation, permitting reversible switching between low- and high-affinity states [54]. Separately, phosphorylation of an IDR in the SWI/SNF subunit DPF3a enhances its association with a structured domain in the H3K36me3 reader HRP2 (HDGFL2), which facilitates recruitment of SWI/SNF complexes to actively expressed target genes, thereby acting as a sensor that modulates SWI/SNF localization in response to differentiation signals [50].
IDRs with multiple PTM sites can also be modified to provide a graded signaling response. For example, the disordered transactivation domain of p53 increases affinity for CBP/p300 with each successive phosphorylation event, resulting in a spectrum of p53 activity [56,57]. PTMs regulating IDRs can also act as repressive marks, such as phosphorylation of an autoinhibitory IDR in the FACT complex, which impairs nucleosome binding [58,59]. Regulated switching of PTM state in IDRs can thus confer substantial functional plasticity.
Degrons and degron masking
IDRs known as degrons control protein stability by mediating regulated proteolysis (Figure 2). E3 ubiquitin ligase-mediated proteasomal degradation of degron-linked proteins has been associated with LLPS through the family of Cullin3-RING E3 ubiquitin ligases and their substrate adaptor SPOP, both of which participate in cellular reaction “hotspots” enriched with ubiquitination activity [60]. SPOP is composed of folded domains that form higher-order oligomers and provide multivalent interfaces for disordered degron-containing substrates [60]. SPOP accumulation in LLPS condensates is disrupted by loss-of-function cancer mutations, which interfere with SPOP phase separation, colocalization with degron-containing substrates and ubiquitination activity in cells [61]. However, not all SPOP substrates phase separate with SPOP [62], which prompts the question of how LLPS influences substrate prioritization.
Disordered degrons are present in many TFs and constitute a widely employed feature governing TF stability [63]. In TFs such as MYC, p53, GCN4, or E2F-1, individual E3 recognition degron sequences are regulated by phosphorylation and these phospho-degrons overlap with disordered transactivation domains [63]. However, conceptual and mechanistic explanations of how such regulatory modules balance activation and degradation have remained elusive. Such a balance is crucial for efficient control of gene expression patterns by these TFs. For example, by limiting MYC and MYCN abundance, disordered phospho-degrons in these TFs help to control release of paused RNAP2 to temporally regulate expression of many genes [64]. Interestingly, switchable IDRs can also mediate degron masking [65], in which interaction with E3 ubiquitin ligases is inhibited by other protein interactions. Switchable IDR-mediated interactions therefore permit either positive or negative control of protein stability.
Multivalent, oligomeric, and higher-order complex assembly
The presence of multiple sites capable of binding interaction partners within an individual protein can enable simultaneous interaction with multiple partner proteins (Figure 2). Such multivalent interactions are essential [23,66] but not exclusive to the establishment of LLPS condensates [67]. IDRs capable of multivalent interactions are found in disordered transactivation domains of TFs, where they promote assembly into transcriptional condensates [26]. Importantly however, the biased composition and clustering of amino acids found in TF transactivation domains [68–70] can also regulate residence time of co-activators in an LLPS-independent manner [67].
IDRs with multiple distinct interaction sites can regulate assembly of protein complexes by serving as selective interaction hubs. One example of a disordered interaction hub is found in IWS1, which influences RNAP2 elongation complex composition and coordinates transcription elongation activities [71]. IWS1 is capable of simultaneous binding to TFIIS, LEDGF, and PPP1R10 via three non-overlapping motifs found within its IDR. Mutation of a single binding site within the IWS1 IDR induces defects in RNAP2 elongation dynamics [71].
Separately, the tumor suppressor p53 harbors two distinct IDR hubs: an N-terminal transactivation domain and C-terminal domain, each of which is composed of multiple distinct interaction sites. These hubs interact with diverse transcription regulators [72], including MDM2 [73], TFIID [74], TFIIH [75], Mediator [76] and others. The direct interaction of p53 with multiple components of TFIID [77,78] and TFIIH [75,77–79] complexes induces DNA-binding conformations at target promoters [74], which guides the assembly of preinitiation complex. Similar examples of multivalent or oligomeric assembly have been reported in other TFs [26,80], regulators of nucleophosmin [81], as well as factors that contribute to DNA damage repair [82].
Functional elements in IDRs that contribute to selectivity
Low-complexity regions (LCRs)
Disordered proteins frequently harbor low-complexity regions (LCRs), which are characterized by compositionally biased, often-repetitive amino acid residue content (Figure 3A). LCRs were originally proposed to be fully unstructured, flexible linkers between structured protein domains [83], however, they are capable of forming secondary structures (e.g. α-helices and β-sheets) [84]. Importantly, LCRs often contribute to protein compartmentalization into membrane-less organelles through LLPS.
Figure 3. Major functional elements found within the IDRs of chromatin and transcription regulators.
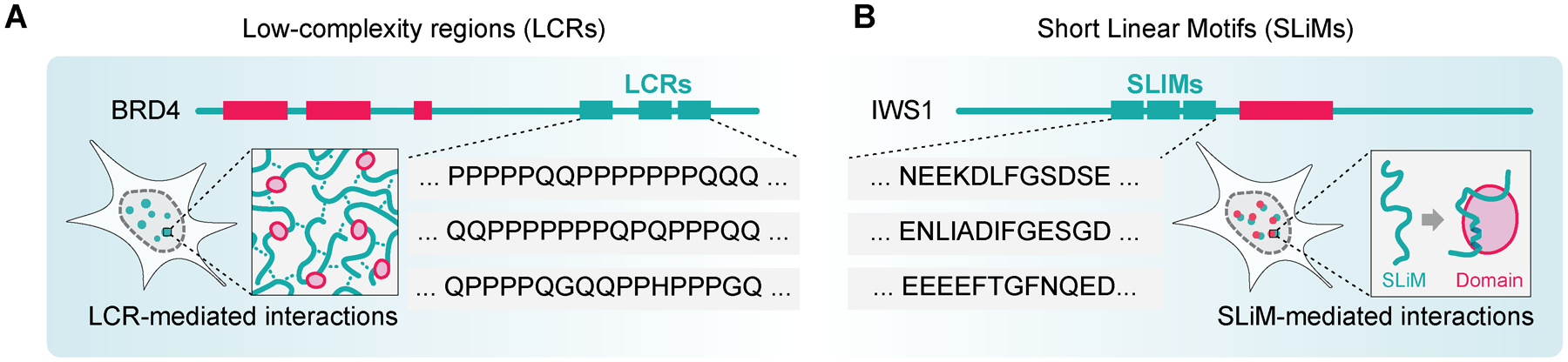
(A) Low-complexity regions (LCRs) form dynamic, non-stoichiometric networks. (B) Short Linear Motifs (SLiMs) mediate selective bipartite interactions with folded domains. IDRs in proteins possess a fluid, non-mutually exclusive spectrum of properties that span from LCR-like to SLiM-like.
Multiple copies of the same LCR can increase the valency of a protein [85]. Indeed, many proteins contain extensive LCR repeats [86,87] that support LLPS by non-stoichiometric multivalent interactions [88]. Such interactions are typically mediated by electrostatic, hydrophobic, cation-π, or π-π contacts [89]. The ability of LCRs to undergo LLPS is dependent on the composition as well as patterning/distribution of amino acids throughout the IDR [90], for example, via alternating segments of aromatic/hydrophobic or charged residues independent of the precise amino acid sequence [90,91]. Even though LCRs are sufficient to induce formation of LLPS condensates [33,92], exactly how LCR features contribute to selectivity among different types of condensates remains an active area of investigation.
Short linear motifs (SLiMs) mediate specific bipartite interactions
Disordered Short Linear Motifs (SLiMs [93]), also known as Eukaryotic Linear Motifs (ELMs [94]) or miniMotifs [95] and related features termed Molecular Recognition Features (MoRFs [96]), support defined and selective protein interactions and associated activities. Here we focus on SLiMs, since they represent minimal and common elements in IDRs that mediate selective interactions [97]. Approximately 78% of known SLiMs are ≤8 amino acid residues in length, but SLiMs up to 23 residues have been documented [93]. SLiMs possess elevated evolutionary conservation even outside of folded regions, and are characterized by selective bipartite interactions with structured protein partners (Figure 3B) [98]. Although SLiMs lack stable 3D structure in solution, they adopt relatively constrained conformations upon binding to their interaction scaffolds (Figure 3B) [98]. Selective interactions mediated by SLiMs range from stable and static to “fuzzy” and dynamic, where the SLiM fluctuates across an ensemble of states [99,100].
Interactions mediated by individual SLiMs are typically micromolar in affinity [93], but some SLiMs have KD values in the low nanomolar range [101]. Furthermore, affinities can be increased by posttranslational regulation, including phosphorylation, SUMOylation, ubiquitination, or others [98,102]. The selectivity of binary interactions between SLiMs and their interaction scaffolds arises from compatibility of IDR residues with the available surfaces on folded interaction partners [103]. SLiMs bind their interaction scaffolds through hydrophobic contacts, electrostatic interactions, hydrogen bonding, and Van der Waals forces [93,104]; hence the large parameter space for distinct interaction surfaces affords strong selectivity for SLiM-mediated interactions.
The spectrum of properties between LCRs and SLiMs
Interestingly, some LCRs possess SLiM-like properties and mediate selective bipartite interactions with folded domains. One of the best-characterized examples of these is the RNAP2 CTD. The unstructured CTD of RNAP2 contains YSPTSPS heptad repeats and thus shows LCR-like compositional bias. On the other hand, structures of the CTD with different binding partners reveal the CTD selectively molds to the folded domains of its binding partners [105,106] and thus has similar properties as SLiMs. Therefore, the RNAP2 CTD is arguably both an LCR and a SLiM, which selectively switches to regulate activity throughout the transcription cycle [107,108]. Similarly, LCRs in transactivation domains of DNA-binding TFs [109] or FG repeats in nucleoporins [110] can also selectively interact with folded domains. The existence of IDRs that have features of both LCRs and SLiMs illustrates that IDRs exist along a fluid spectrum of properties that vary between SLiM-like and LCR-like, and that these properties are not necessarily mutually exclusive.
Avenues for SLiMs to contribute to LLPS specificity
SLiMs make important contributions to soluble protein-protein interactions, and their ability to selectively bind folded domains can also be expected to influence specificity within LLPS condensates. One path by which SLiMs regulate LLPS is the nucleation of condensates. Indeed, arrays containing repeats of single SLiMs induce demixing phase transitions in the presence of their interaction domains [23,66,111]. Moreover, SLiMs regulated by PTMs may guide the composition of condensates through signal-dependent interactions. SLiM-mediated interactions may furthermore influence subnuclear localization of condensates by mediating proximity with folded domains found at important features (e.g. chromatin reading domains, or DNA binding domains). Below, we review examples of domains that read disordered SLiMs with specificity.
Structured protein domains that serve as interaction platforms for IDRs in transcription
Many proteins employ recurrent folds to selectively bind IDRs. Here we highlight several examples of such folds, including the TFIIS N-terminal domain (TND), αα-hub, KIX domain, transcriptional adapter zinc finger (TAZ) domain, WD40 repeat (WDR), as well as PDZ, WW, SH2 and SH3 domains. Despite representing a small portion of the interaction modules that recognize disordered protein, these families illustrate the high degree of structural diversity among such interaction modules. Each of these domain families interacts with SLiMs through distinct structural configurations (Figure 4). In each of the above cases, the domains themselves are not known to possess enzymatic activity, further supporting protein scaffolding as their primary function.
Figure 4. Examples of SLiM interaction scaffolds in chromatin and transcription regulators.
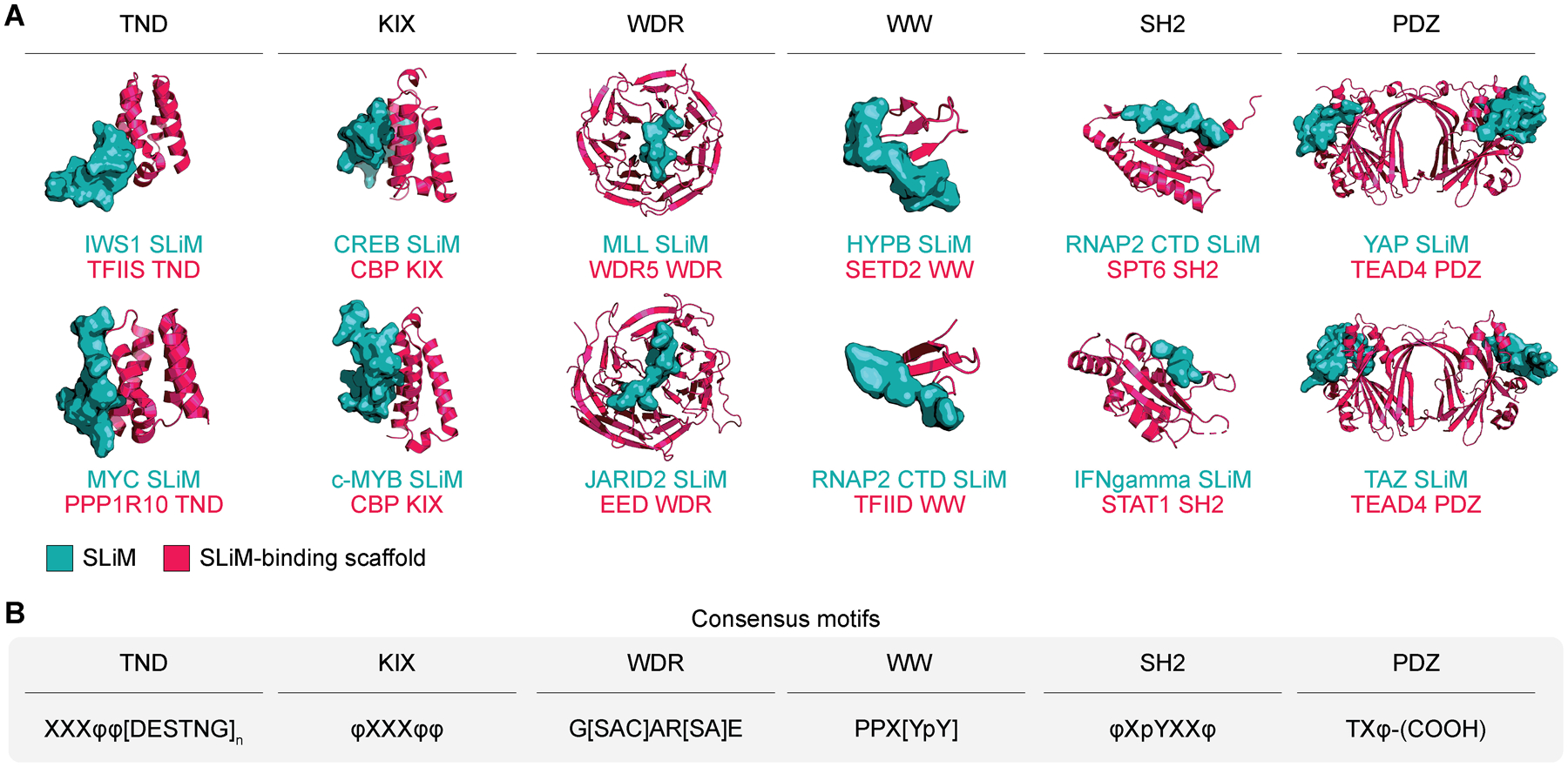
(A) SLiMs (teal surface) are depicted in complexes with their interaction scaffolds (fuchsia ribbon). SLiM-mediated interactions employ recurrent structural configurations (PDB accessions 6ZV4, 7LQT, 1KDX, 1SB0, 3P4F, 5HYN, 2MDJ, 1F8A, 5VKO, 1YVL, 3JUA, 5GN0). (B) Examples of SLiM consensus sequences for SLiM interaction scaffolds. Individual amino acids are represented by their IUPAC codes. X denotes any amino acid, φ represents hydrophobic amino acid; p-prefix indicates phosphorylated amino acid; square brackets represent any one of the amino acids contained within the brackets; subscript n denotes repetition; (COOH) denotes carboxy terminus.
Examples of SLiM-binding domains that are primarily composed of α-helices include TNDs, αα-hubs, KIX domains, and TAZ domains. The TND is a small five-helix bundle (Figure 4A) and the most significantly enriched fold among the transcription elongation machinery [71]. TNDs are recognized by a family of conserved SLiMs called TND-interacting motifs (TIMs). TIMs are composed of bulky hydrophobic residues with α-helical propensity followed by a negatively charged stretch and optional FXGF motif (consensus motif in Figure 4B). The TND-TIM module is found in TFs [112], SWI/SNF chromatin remodeling complexes [50], as well as other members of transcription machinery, including COMPASS, Super Elongation Complex, TFIIS, and many others [71]. The αα-hub consists of 3 to 5 α-helices that form an αα-hairpin. Most SLiMs interact with αα-hubs through bulky residues that contact the hydrophobic αα-hub cleft. The αα-hub is found in many transcription regulators, such as TAF4, where it interacts with SLiMs from multiple transcriptional regulators [113], or in CREB binding protein, where it supports the interaction with a SLiM in p53 [114]. The multivalent interaction between CREB binding protein and p53 is also mediated by two TAZ domains in CREB binding protein, which recognize two distinct SLiMs in p53 [49]. KIX domains are composed of three α- and two 310-helices that provide two distinct SLiM-binding surfaces. KIX domains are recognized by bulky hydrophobic residues from SLiMs in the disordered transactivation domains of many TFs. For example, the CBP/p300 KIX domain is the primary interaction site for numerous TFs and other regulators, including CREB, c-Myb, KMT2A, c-Jun, E2A, and FOXO3 [57,115,116]. The high versatility of α-helix scaffolds as interaction platforms is also illustrated by the helical portions of ordered domains from many nuclear receptors that selectively bind the NR box (or LXXLL and related motifs) [117].
WDR is one of the most abundant domains in the eukaryotic proteome, and proteins containing this domain account for ~1% of the human proteome [118]. WDRs are organized as repeats of the WD40 β-propeller fold, which binds highly diverse SLiMs through multiple interaction sites. The most common interaction site is the top central tunnel of the WDR domain (Figure 4) [118]. WDR domains regulate wide-ranging activities, including the cell cycle, apoptosis, signal transduction, and are present in many transcriptional regulators, including the TAF5 subunit of TFIID, the MED16 subunit of Mediator, or RBBP4 and EED, both components of Polycomb repressive complex 2 (PRC2) [119]. SLiMs that recognize this domain are found in the MLL (KMT2) family of histone methyltransferases, which binds to WDR5 through the WDR5 interaction (WIN) consensus motif [51].
Domains that bind unstructured proline-rich motifs include WW and SH3 domains. WW domains fold into a three-stranded antiparallel β sheet that selectively bind proline-rich SLiMs [120]. WW domains can be found in iso-prolyl isomerase PIN1 and E3 ubiquitin ligase NEDD4, both of which bind the disordered RNAP2 CTD. Additionally, the conserved WW domain in the H3K36me3 writer SETD2 plays an autoinhibitory role by interacting with a polyproline stretch at the SETD2 C-terminus [121]. The SH3 domain is a structurally distinct fold that like the WW domain interacts with proline-rich motifs. SH3 domains consist of five or six β-strands arranged as two tightly packed anti-parallel β-sheets, and are found in many tyrosine kinases, including those in the ABL [122] and SRC families [123].
PDZ and SH2 domains, as well as their interaction motifs, are enriched among transcription regulators involved in signaling. While SH2 domains are prominent scaffolds that bind switchable phosphotyrosine-containing SLiMs, PDZ domains recognize several classes of very short degenerate C-terminal SLiMs [94]. Examples of such interactions are found in TAZ [124] and STAT [125] TF families, which employ these domains for transcriptional activation. The histone chaperone SPT6 furthermore employs its SH2 domain for recruitment to Ser-2-phosphorylated RNAP2 [126]. Deeper examination of these SLiM-mediated interactions may reveal new mechanisms underlying selective signal-dependent transcription activation.
In addition to the functions described above (Figure 2), folded domains that recognize SLiMs are also frequently hijacked by viruses. For example, lentiviruses including HIV-1 employ viral mimicry of a SLiM capable of binding TNDs on H3K36me3 readers to integrate into transcriptionally active regions of chromatin [127]. Similarly, the TAZ2 domain of the histone acetyltransferase CBP/p300 family is hijacked by a conserved disordered motif in the adenoviral family protein E1A [128]. The ET domain conserved among the BET family, including in BRD4, is also hijacked by a viral motif conserved among ɣ-retroviral integrases [129]. Furthermore, a motif mimicking the disordered N-terminal tail of histone H3 in the NS1 protein of influenza coopts the tandem chromodomains of chromatin remodeler CHD1 [130]. The frequent utilization of SLiM-like sequences among endogenous and viral factors suggests that controlling these interactions may represent powerful opportunities for therapeutic intervention.
Targeting disorder in transcriptional regulators for therapeutic benefit
IDR-mediated interactions are associated with disease, including cancer, neurodegenerative, and inflammatory disorders [131–134], which raises the possibility of modulating these interactions for therapeutic benefit. In many cases, molecules that target LLPS condensate formation also inhibit other selective functions of their target proteins (e.g. interfering with post-translation modifiers of LCRs that mediate LLPS) [135–138], and hence many questions remain regarding the sufficiency of LLPS per se as a therapeutic target.
Several approaches interfering with SLiM-mediated interactions have had considerable preliminary success as therapeutic strategies. One approach is design of small molecules that outcompete SLiMs for interaction with SLiM-binding scaffolds. For example, inhibitors of the MLL-Menin interface [139,140] have been successfully employed in pre-clinical studies and are currently in clinical trials to target acute leukemia. SLiMs or SLiM-binding scaffolds can also be degraded by targeting any druggable part of the full-length protein using PROTACs [141–143]. In addition, the development of small molecules that stabilize SLiM-mediated interactions [144] suggests that stabilization of such contacts by molecular glues may also be a useful therapeutic paradigm.
Despite the conventional expectation that IDRs have poor ligand selectivity profiles, a small molecule targeting the disordered transactivation domain of androgen receptor was recently reported [145]. Separately, a unique targetable site within an IDR of MYC identified through a cysteine-reactive covalent ligand was successfully developed with efficacy against breast cancer [146]. Such results suggest that IDRs can be selectively targeted and that covalent ligands may represent a significant breakthrough for direct targeting of IDRs.
Enzymes that regulate SLiMs via PTMs are also potential therapeutic targets. For example, casein kinase 2 (CK2), a serine/threonine kinase, modulates the affinities of many SLiM-mediated interactions via phosphorylation [54,147], and CK2 inhibitors are currently in clinical trials for hematologic and solid tumors [148]. These efforts illustrate that both direct and indirect targeting of SLiMs can be a viable and potent therapeutic strategy.
Concluding remarks
The explosion of interest in disordered protein has prompted extensive dialog about how IDRs achieve specificity to exert distinct biological functions. While the principles that govern specificity underlying LLPS are still emerging, recent discoveries demonstrate that a substantial degree of IDR-mediated specificity arises via contacts between SLiMs and folded proteins. Because of the selective interactions described above, SLiMs can be expected to confer selectivity to several properties of phase separated condensates. Therefore, exploring the contributions of SLiMs towards the nucleation, localization, or composition of condensates represents a compelling scientific direction (see Outstanding questions).
Outstanding Questions Box:
What aspects of chromatin and transcription are regulated by intrinsically disordered regions (IDRs)?
What roles do short linear motifs (SLiMs) play in liquid-liquid phase separation (LLPS) and how they contribute to selective partitioning of proteins into different types of condensates?
What are the best approaches for high-throughput identification of selective interactions maintained by IDRs, as well as for characterization of their functions?
What is the grammar of disordered interactions and how do cells integrate signals from multiple IDRs?
Although IDRs play important roles in transcription regulation, they are greatly understudied compared to folded domains. Therefore, development of new tools that permit high-throughput examination of IDR biology in the cellular context of their native binding partners represents an important goal. Moreover, as described above, pharmaceutical targeting of these features may also be more feasible than previously assumed. Because the human proteome is estimated to contain hundreds of thousands of SLiM instances [102], it is almost certain that many more selective protein interaction modules encoded in the IDRs of transcription regulators are waiting to be discovered.
Box 2. SLiM biology represents a major frontier that spans biomedical disciplines.
Current estimates propose that there may be over a million instances of short functional peptide motifs in the human proteome [102]. However, only ~2,000 of these are annotated and SLiMs represent a major, but understudied class among eukaryotic regulatory modules [102]. Given the important and selective roles that these known short peptides regulate, their research holds great opportunities for new discoveries. SLiM biology also has significant implications for human health. SLiMs are often involved in aberrant signaling in cancer [157] and their plasticity is exploited by pathogens, such as viruses, which often mimic peptide motifs of host cell interactions to hijack cellular pathways [158]. In addition, mutations disrupting these short peptides as well as mutations of proteins that regulate their PTMs are known disease drivers [159]. Therefore, their protein-protein interaction networks represent an unexplored supply of potential new therapeutic targets. Another major utility of the current knowledge of SLiM biology is in cellular engineering for driving fundamental discoveries in biomedical research. For example, many labs already use SLiMs to direct sub-cellular localization (e.g. nuclear localization signals [48]), for engineering of targeted protein degradation (e.g. auxin-induced degron (AID) [160]), or to change integration site selection of viruses to engineer safer viral vectors for gene therapy [161]. Due to their small size, modular function, and diverse roles, SLiMs may represent powerful building blocks for the study of fundamental cellular biology as well as for synthetic biology, where minor edits can alter function in many ways. Given the broad roles of SLiMs, many opportunities for their application across disciplines from basic research to engineering can be envisioned.
Highlights:
Transcription and chromatin regulation represent the most highly enriched cellular processes containing high disordered protein content.
IDRs harbor two types of sequences with documented regulatory roles: Short linear motifs (SLiMs) and low-complexity regions (LCRs).
IDRs exist along a spectrum of properties that varies between SLiM-like and LCR-like, and these properties are not necessarily mutually exclusive.
LCR- and SLiM-mediated interactions contribute to selective partitioning and distinct composition of phase separated condensates.
IDRs remain understudied, but have important contributions to human health and are druggable.
Acknowledgements
We thank members of the Hodges lab for helpful feedback during manuscript preparation. This work was supported by grants from the NIH (R35GM137996 to H.C.H.) and the Cancer Prevention and Research Institute of Texas (RR170036 to H.C.H.).
Glossary:
-
αα-hub
SLiM interaction scaffold composed of α-helices that binds SLiMs enriched in hydrophobic residues
Cysteine-reactive covalent ligand
small molecule designed to selectively interact with a cysteine residue on proteins of interest through a covalent bond
Degron
element within a protein that is sufficient for targeting the protein to E3 ubiquitin ligases for ubiquitin-mediated proteasomal degradation
Intrinsically disordered region (IDR)
polypeptide segment in proteins that is natively unstructured and adopts heterogeneous conformations. In the absence of interaction partners, IDRs lack stable three-dimensional structure
KID-interacting (KIX) domain
interaction scaffold that binds IDRs named kinase-inducible domains (KIDs). The KIX domain is composed of α-helices that bind bulky hydrophobic residues in KIDs
Low-complexity region (LCR)
disordered protein region whose content is enriched in repeats of a small number of amino acid residues. LCRs typically have sequence composition that is strongly biased in amino acid content
Liquid-liquid phase separation (LLPS)
phenomenon resulting in partitioning of two or more liquid components into distinct liquid phases
Molecular Recognition Features (MoRFs)
10–70 amino acid segments within IDRs that undergo disorder-to-order transitions to mediate specific interactions
NR box
a disordered sequence composed of LXXLL, FXXLL, or related motifs that is recognized by α-helical regions in nuclear receptors
SH2 domain
SLiM interaction scaffold composed of a characteristic fold containing both α-helices and β-strands that recognizes phosphotyrosine-containing motifs
SH3 domain
SLiM interaction scaffold composed of a β-barrel fold that recognizes proline-rich motifs
Short linear motif (SLiM)
also known as a Eukaryotic Linear Motif (ELM) or miniMotif, this disordered short polypeptide segment is typically 3–10 amino acids long and mediates selective protein-protein interactions with SLiM interaction scaffolds
Transcription Adaptor putative Zinc finger (TAZ) domain
SLiM-interaction scaffold that forms an α-helical fold
TFIIS N-terminal domain (TND)
SLiM interaction scaffold composed of a five-helix bundle that recognizes TIMs
TND-interacting motif (TIM)
SLiM that selectively recognizes a TND domain
WD40 domain
conserved domain characterized by a four-stranded anti-parallel β-sheet structure that represents the basic unit of a WDR domain
WD40 repeat (WDR)
SLiM interaction scaffold composed of tandem repeats of WD40 domains that typically forms circularized solenoid-like structure. WDRs bind diverse SLiMs, including WINs
WDR5 interaction (WIN) motif
SLiM that selectively recognizes WDRs
WW domain
SLiM interaction scaffold composed of a triple-stranded β-sheet that recognizes proline-rich motifs
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
None are declared by the authors.
References
- 1.Dyson HJ and Wright PE (2021) NMR illuminates intrinsic disorder. Curr. Opin. Struct. Biol 70, 44–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kosol S et al. (2013) Structural characterization of intrinsically disordered proteins by NMR spectroscopy. Molecules 18, 10802–10828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oates ME et al. (2013) D2P2: Database of disordered protein predictions. Nucleic Acids Res. 41, D508–D516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao B et al. (2021) DescribePROT: Database of amino acid-level protein structure and function predictions. Nucleic Acids Res. 49, D298–D308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jumper J et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xue B et al. (2010) PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta - Proteins Proteomics 1804, 996–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sigler PB (1988) Acid blobs and negative noodles. Nature 333, 210–212 [DOI] [PubMed] [Google Scholar]
- 8.Tompa P et al. (2015) Intrinsically disordered proteins: Emerging interaction specialists. Curr. Opin. Struct. Biol 35, 49–59 [DOI] [PubMed] [Google Scholar]
- 9.Sabari BR et al. (2020) Biomolecular Condensates in the Nucleus. Trends Biochem. Sci 45, 961–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rippe K (2022) Liquid–Liquid Phase Separation in Chromatin. Cold Spring Harb. Perspect. Biol 14, a040683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su Q et al. (2021) Liquid-liquid phase separation: Orchestrating cell signaling through time and space. Mol. Cell 81, 4137–4146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie H et al. (2007) Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J. Proteome Res 6, 1882–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson PJ et al. (2016) Structure of a Complete Mediator-RNA Polymerase II Pre-Initiation Complex. Cell 166, 1411–1422.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Louder RK et al. (2016) Structure of promoter-bound TFIID and model of human preinitiation complex assembly. Nature 531, 604–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernecky C et al. (2017) Structure of a transcribing RNA polymerase II-DSIF complex reveals a multidentate DNA-RNA clamp. Nat. Struct. Mol. Biol 24, 809–815 [DOI] [PubMed] [Google Scholar]
- 16.Vos SM et al. (2020) Structure of complete Pol II–DSIF–PAF–SPT6 transcription complex reveals RTF1 allosteric activation. Nat. Struct. Mol. Biol 27, 668–677 [DOI] [PubMed] [Google Scholar]
- 17.He S et al. (2020) Structure of nucleosome-bound human BAF complex. Science 367, 875–881 [DOI] [PubMed] [Google Scholar]
- 18.Liu Y et al. (2020) FACT caught in the act of manipulating the nucleosome. Nature 577, 426–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farnung L et al. (2018) Structure of transcribing RNA polymerase II-nucleosome complex. Nat. Commun 9, 5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Filipovski M et al. (2022) Structural basis of nucleosome retention during transcription elongation. Science 376, 1313–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banani SF et al. (2017) Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol 18, 285–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hnisz D et al. (2017) A Phase Separation Model for Transcriptional Control. Cell 169, 13–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibson BA et al. (2019) Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 179, 470–484.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McSwiggen DT et al. (2019) Evaluating phase separation in live cells: diagnosis, caveats, and functional consequences. Genes Dev. 33, 1619–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riback JA et al. (2020) Composition-dependent thermodynamics of intracellular phase separation. Nature 581, 209–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boija A et al. (2018) Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 175, 1842–1855.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai D et al. (2019) Phase separation of YAP reorganizes genome topology for long-term YAP target gene expression. Nat. Cell Biol 21, 1578–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu Y et al. (2020) Phase separation of TAZ compartmentalizes the transcription machinery to promote gene expression. Nat. Cell Biol 22, 453–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sabari BR et al. (2018) Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho WK et al. (2018) Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo YE et al. (2019) Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 572, 543–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu H et al. (2018) Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature 558, 318–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boehning M et al. (2018) RNA polymerase II clustering through carboxy-terminal domain phase separation. Nat. Struct. Mol. Biol 25, 833–840 [DOI] [PubMed] [Google Scholar]
- 34.Quintero-Cadena P et al. (2020) RNA Pol II Length and Disorder Enable Cooperative Scaling of Transcriptional Bursting. Mol. Cell 79, 207–220.e8 [DOI] [PubMed] [Google Scholar]
- 35.Kwon I et al. (2013) Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell 155, 1049–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cramer P (2019) Organization and regulation of gene transcription. Nature 573, 45–54 [DOI] [PubMed] [Google Scholar]
- 37.Hathaway NA et al. (2012) Dynamics and memory of heterochromatin in living cells. Cell 149, 1447–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Verschure PJ et al. (2005) In Vivo HP1 Targeting Causes Large-Scale Chromatin Condensation and Enhanced Histone Lysine Methylation. Mol. Cell. Biol 25, 4552–4564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Canzio D et al. (2011) Chromodomain-mediated oligomerization of HP1 suggests a nucleosome-bridging mechanism for heterochromatin assembly. Mol. Cell 41, 67–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keenen MM et al. (2021) HP1 proteins compact dna into mechanically and positionally stable phase separated domains. Elife 10, e64563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larson AG et al. (2017) Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547, 236–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanulli S et al. (2019) HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature 575, 390–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strom AR et al. (2017) Phase separation drives heterochromatin domain formation. Nature 547, 241–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erdel F et al. (2020) Mouse Heterochromatin Adopts Digital Compaction States without Showing Hallmarks of HP1-Driven Liquid-Liquid Phase Separation. Mol. Cell 78, 236–249.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bosch-Presegué L et al. (2017) Mammalian HP1 Isoforms Have Specific Roles in Heterochromatin Structure and Organization. Cell Rep. 21, 2048–2057 [DOI] [PubMed] [Google Scholar]
- 46.Aucott R et al. (2008) HP1 -β is required for development of the cerebral neocortex and neuromuscular junctions. J. Cell Biol 183, 597–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shin Y et al. (2018) Liquid Nuclear Condensates Mechanically Sense and Restructure the Genome. Cell 175, 1481–1491.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu J et al. (2021) Types of nuclear localization signals and mechanisms of protein import into the nucleus. Cell Commun. Signal 19, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krois AS et al. (2016) Recognition of the disordered p53 transactivation domain by the transcriptional adapter zinc finger domains of CREB-binding protein. Proc. Natl. Acad. Sci. U. S. A 113, E1853–E1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu X et al. (2020) HRP2-DPF3a-BAF complex coordinates histone modification and chromatin remodeling to regulate myogenic gene transcription. Nucleic Acids Res. 48, 6563–6582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaustov L et al. (2019) The MLL1 trimeric catalytic complex is a dynamic conformational ensemble stabilized by multiple weak interactions. Nucleic Acids Res. 47, 9433–9447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Han X et al. (2020) Roles of the BRD4 short isoform in phase separation and active gene transcription. Nat. Struct. Mol. Biol 27, 333–341 [DOI] [PubMed] [Google Scholar]
- 53.Tesina P et al. (2015) Multiple cellular proteins interact with LEDGF/p75 through a conserved unstructured consensus motif. Nat. Commun 6, 7968. [DOI] [PubMed] [Google Scholar]
- 54.Sharma S et al. (2018) Affinity switching of the LEDGF/p75 IBD interactome is governed by kinase-dependent phosphorylation. Proc. Natl. Acad. Sci. U. S. A 115, E7053–E7062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Čermaková K et al. (2014) Validation and structural characterization of the LEDGF/p75-MLL interface as a new target for the treatment of MLL-dependent leukemia. Cancer Res. 74, 5139–5151 [DOI] [PubMed] [Google Scholar]
- 56.Lee CW et al. (2010) Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation. Proc. Natl. Acad. Sci. U. S. A 107, 19290–19295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wright PE and Dyson HJ (2015) Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol 16, 18–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hashimoto M et al. (2013) Phosphorylation-coupled intramolecular dynamics of unstructured regions in chromatin remodeler FACT. Biophys. J 104, 2222–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aoki D et al. (2020) Ultrasensitive Change in Nucleosome Binding by Multiple Phosphorylations to the Intrinsically Disordered Region of the Histone Chaperone FACT. J. Mol. Biol 432, 4637–4657 [DOI] [PubMed] [Google Scholar]
- 60.Marzahn MR et al. (2016) Higher-order oligomerization promotes localization of SPOP to liquid nuclear speckles. EMBO J. 35, 1254–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bouchard JJ et al. (2018) Cancer Mutations of the Tumor Suppressor SPOP Disrupt the Formation of Active, Phase-Separated Compartments. Mol. Cell 72, 19–36.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Usher ET et al. (2021) Intrinsically disordered substrates dictate SPOP subnuclear localization and ubiquitination activity. J. Biol. Chem 296, 100693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Salghetti SE et al. (2000) Functional overlap of sequences that activate transcription and signal ubiquitin-mediated proteolysis. Proc. Natl. Acad. Sci. U. S. A 97, 3118–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baluapuri A et al. (2020) Target gene-independent functions of MYC oncoproteins. Nat. Rev. Mol. Cell Biol 21, 255–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guharoy M et al. (2022) Degron masking outlines degronons, co-degrading functional modules in the proteome. Commun. Biol 5, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li P et al. (2012) Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Trojanowski J et al. (2022) Transcription activation is enhanced by multivalent interactions independent of phase separation. Mol. Cell 82, 1878–1893.e10 [DOI] [PubMed] [Google Scholar]
- 68.Erijman A et al. (2020) A High-Throughput Screen for Transcription Activation Domains Reveals Their Sequence Features and Permits Prediction by Deep Learning. Mol. Cell 78, 890–902.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ravarani CN et al. (2018) High-throughput discovery of functional disordered regions: investigation of transactivation domains. Mol. Syst. Biol 14, e8190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tycko J et al. (2020) High-Throughput Discovery and Characterization of Human Transcriptional Effectors. Cell 183, 2020–2035.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cermakova K et al. (2021) A ubiquitous disordered protein interaction module orchestrates transcription elongation. Science 374, 1113–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Uversky VN (2016) p53 proteoforms and intrinsic disorder: An illustration of the protein structure–function continuum concept. Int. J. Mol. Sci 17, 1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kussie PH et al. (1996) Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274, 948–953 [DOI] [PubMed] [Google Scholar]
- 74.Coleman RA et al. (2017) p53 Dynamically Directs TFIID Assembly on Target Gene Promoters. Mol. Cell. Biol 37, e00085–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Di Lello P et al. (2006) Structure of the Tfb1/p53 Complex: Insights into the Interaction between the p62/Tfb1 Subunit of TFIIH and the Activation Domain of p53. Mol. Cell 22, 731–740 [DOI] [PubMed] [Google Scholar]
- 76.Meyer KD et al. (2010) P53 activates transcription by directing structural shifts in Mediator. Nat. Struct. Mol. Biol 17, 753–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thut CJ et al. (1995) P53 transcriptional activation mediated by coactivators TAFII40 and TAFII60. Science 267, 100–104 [DOI] [PubMed] [Google Scholar]
- 78.Lu H and Levine AJ (1995) Human TAFII31 protein is a transcriptional coactivator of the p53 protein. Proc. Natl. Acad. Sci. U. S. A 92, 5154–5158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen X et al. (2021) Structural insights into preinitiation complex assembly on core promoters. Science 372, eaba8490 [DOI] [PubMed] [Google Scholar]
- 80.Warfield L et al. (2014) A sequence-specific transcription activator motif and powerful synthetic variants that bind Mediator using a fuzzy protein interface. Proc. Natl. Acad. Sci. U. S. A 111, E3506–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mitrea DM et al. (2014) Structural polymorphism in the N-terminal oligomerization domain of NPM1. Proc. Natl. Acad. Sci. U. S. A 111, 4466–4471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Clark S et al. (2018) Multivalency regulates activity in an intrinsically disordered transcription factor. Elife 7, e36258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huntley MA and Golding GB (2002) Simple sequences are rare in the Protein Data Bank. Proteins Struct. Funct. Genet 48, 134–140 [DOI] [PubMed] [Google Scholar]
- 84.Kumari B et al. (2015) Low complexity and disordered regions of proteins have different structural and amino acid preferences. Mol. Biosyst 11, 585–594 [DOI] [PubMed] [Google Scholar]
- 85.Schuster BS et al. (2018) Controllable protein phase separation and modular recruitment to form responsive membraneless organelles. Nat. Commun 9, 2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee B et al. (2022) A unified view of low complexity region (LCRs) across species. Elife 11, e77058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kamel M et al. (2019) Repeatability in protein sequences. J. Struct. Biol 208, 86–91 [DOI] [PubMed] [Google Scholar]
- 88.Martin EW and Holehouse AS (2020) Intrinsically disordered protein regions and phase separation: Sequence determinants of assembly or lack thereof. Emerg. Top. Life Sci 4, 307–329 [DOI] [PubMed] [Google Scholar]
- 89.Vernon RMC et al. (2018) Pi-Pi contacts are an overlooked protein feature relevant to phase separation. Elife 7, e31486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pak CW et al. (2016) Sequence Determinants of Intracellular Phase Separation by Complex Coacervation of a Disordered Protein. Mol. Cell 63, 72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lyons H et al. (2023) Functional partitioning of transcriptional regulators by patterned charge blocks. Cell DOI: 10.1016/j.cell.2022.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kato M et al. (2012) Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Davey NE et al. (2012) Attributes of short linear motifs. Mol. Biosyst 8, 268–281 [DOI] [PubMed] [Google Scholar]
- 94.Kumar M et al. (2020) ELM-the eukaryotic linear motif resource in 2020. Nucleic Acids Res. 48, D296–D306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lyon KF et al. (2018) Minimotif Miner 4: A million peptide minimotifs and counting. Nucleic Acids Res. 46, D465–D470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mohan A et al. (2006) Analysis of Molecular Recognition Features (MoRFs). J. Mol. Biol 362, 1043–1059 [DOI] [PubMed] [Google Scholar]
- 97.Van Der Lee R et al. (2014) Classification of intrinsically disordered regions and proteins. Chem. Rev 114, 6589–6631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Van Roey K et al. (2014) Short linear motifs: Ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev 114, 6733–6778 [DOI] [PubMed] [Google Scholar]
- 99.Tompa P and Fuxreiter M (2008) Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci 33, 2–8 [DOI] [PubMed] [Google Scholar]
- 100.Sharma R et al. (2015) Fuzzy complexes: Specific binding without complete folding. FEBS Lett. 589, 2533–2542 [DOI] [PubMed] [Google Scholar]
- 101.O’Shea C et al. (2017) Structures and Short Linear Motif of Disordered Transcription Factor Regions Provide Clues to the Interactome of the Cellular Hub Protein Radical-induced Cell Death 1. J. Biol. Chem 292, 512–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tompa P et al. (2014) A Million peptide motifs for the molecular biologist. Mol. Cell 55, 161–169 [DOI] [PubMed] [Google Scholar]
- 103.Cermakova K et al. (2023) The TFIIS N-terminal domain (TND): A transcription assembly module at the interface of order and disorder. Biochem. Soc. Trans 51, doi: 10.1042/BST20220342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bugge K et al. (2020) Interactions by Disorder – A Matter of Context. Front. Mol. Biosci 7, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fabrega C et al. (2003) Structure of an mRNA capping enzyme bound to the phosphorylated carboxy-terminal domain of RNA polymerase II. Mol. Cell 11, 1549–1561 [DOI] [PubMed] [Google Scholar]
- 106.Verdecia MA et al. (2000) Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat. Struct. Biol 7, 639–643 [DOI] [PubMed] [Google Scholar]
- 107.Davey NE (2019) The functional importance of structure in unstructured protein regions. Curr. Opin. Struct. Biol 56, 155–163 [DOI] [PubMed] [Google Scholar]
- 108.Schier AC and Taatjes DJ (2020) Structure and mechanism of the RNA polymerase II transcription machinery. Genes Dev. 34, 465–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chong S et al. (2018) Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361, eaar2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Otsuka S et al. (2008) Individual binding pockets of importin-β for FG-nucleoporins have different binding properties and different sensitivities to RanGTP. Proc. Natl. Acad. Sci. U. S. A 105, 16101–16106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Harmon TS et al. (2017) Intrinsically disordered linkers determine the interplay between phase separation and gelation in multivalent proteins. Elife 6, e30294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wei Y et al. (2022) The MYC oncoprotein directly interacts with its chromatin cofactor PNUTS to recruit PP1 phosphatase. Nucleic Acids Res. 50, 3505–3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang X et al. (2007) Conserved region I of human coactivator TAF4 binds to a short hydrophobic motif present in transcriptional regulators. Proc. Natl. Acad. Sci. U. S. A 104, 7839–7844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lee CW et al. (2010) Structure of the p53 transactivation domain in complex with the nuclear receptor coactivator binding domain of CREB binding protein. Biochemistry 49, 9964–9971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.De Guzman RN et al. (2006) Structural basis for cooperative transcription factor binding to the CBP coactivator. J. Mol. Biol 355, 1005–1013 [DOI] [PubMed] [Google Scholar]
- 116.Dyson HJ and Wright PE (2016) Role of intrinsic protein disorder in the function and interactions of the transcriptional coactivators CREB-binding Protein (CBP) and p300. J. Biol. Chem 291, 6714–6722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yu X et al. (2020) Structural Insights of Transcriptionally Active, Full-Length Androgen Receptor Coactivator Complexes. Mol. Cell 79, 812–823.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stirnimann CU et al. (2010) WD40 proteins propel cellular networks. Trends Biochem. Sci 35, 565–574 [DOI] [PubMed] [Google Scholar]
- 119.Zou XD et al. (2016) Genome-wide Analysis of WD40 Protein Family in Human. Sci. Rep 6, 39262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Otte L (2003) WW domain sequence activity relationships identified using ligand recognition propensities of 42 WW domains. Protein Sci. 12, 491–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gao YG et al. (2014) Autoinhibitory structure of the WW domain of HYPB/SETD2 regulates its interaction with the proline-rich region of huntingtin. Structure 22, 378–386 [DOI] [PubMed] [Google Scholar]
- 122.Smith KM et al. (2003) Autoinhibition of Bcr-Abl through its SH3 domain. Mol. Cell 12, 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Moroco JA et al. (2014) Differential sensitivity of Src-family kinases to activation by SH3 domain displacement. PLoS One 9, e105629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kanai F et al. (2000) TAZ: A novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 19, 6778–6791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.de Araujo ED et al. (2019) Structural implications of stat3 and stat5 sh2 domain mutations. Cancers (Basel). 11, 1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sdano MA et al. (2017) A novel SH2 recognition mechanism recruits Spt6 to the doubly phosphorylated RNA polymerase II linker at sites of transcription. Elife 6, e28723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cermakova K et al. (2016) Lessons Learned: HIV Points the Way Towards Precision Treatment of Mixed-Lineage Leukemia. Trends Pharmacol. Sci 37, 660–671 [DOI] [PubMed] [Google Scholar]
- 128.Pelka P et al. (2008) Intrinsic Structural Disorder in Adenovirus E1A: a Viral Molecular Hub Linking Multiple Diverse Processes. J. Virol 82, 7252–7263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.DeRijck J et al. (2013) The BET Family of Proteins Targets Moloney Murine Leukemia Virus Integration near Transcription Start Sites. Cell Rep. 5, 886–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Marazzi I et al. (2012) Suppression of the antiviral response by an influenza histone mimic. Nature 483, 428–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ahn JH et al. (2021) Phase separation drives aberrant chromatin looping and cancer development. Nature 595, 591–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zbinden A et al. (2020) Phase Separation and Neurodegenerative Diseases: A Disturbance in the Force. Dev. Cell 55, 45–68 [DOI] [PubMed] [Google Scholar]
- 133.Du M and Chen ZJ (2018) DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, 704–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li CH et al. (2020) MeCP2 links heterochromatin condensates and neurodevelopmental disease. Nature 586, 440–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mao YS et al. (2011) Direct visualization of the co-transcriptional assembly of a nuclear body by noncoding RNAs. Nat. Cell Biol 13, 95–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lu B et al. (2021) Pharmacological Inhibition of Core Regulatory Circuitry Liquid–liquid Phase Separation Suppresses Metastasis and Chemoresistance in Osteosarcoma. Adv. Sci 8, e2101895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Düster R et al. (2021) 1,6-Hexanediol, commonly used to dissolve liquid-liquid phase separated condensates, directly impairs kinase and phosphatase activities. J. Biol. Chem 296, 100260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Klein IA et al. (2020) Partitioning of cancer therapeutics in nuclear condensates. Science 368, 1386–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Grembecka J et al. (2012) Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat. Chem. Biol 8, 277–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Krivtsov AV et al. (2019) A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 36, 660–673.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Békés M et al. (2022) PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov 21, 181–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Samarasinghe KTG and Crews CM (2021) Targeted protein degradation: A promise for undruggable proteins. Cell Chem. Biol 28, 934–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cermakova K and Courtney Hodges H (2018) Next-generation drugs and probes for chromatin biology: From targeted protein degradation to phase separation. Molecules 23, 1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sijbesma E et al. (2020) Structure-based evolution of a promiscuous inhibitor to a selective stabilizer of protein–protein interactions. Nat. Commun 11, 3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sadar MD (2020) Discovery of drugs that directly target the intrinsically disordered region of the androgen receptor. Expert Opin. Drug Discov 15, 551–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Boike L et al. (2021) Discovery of a Functional Covalent Ligand Targeting an Intrinsically Disordered Cysteine within MYC. Cell Chem. Biol 28, 4–13.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Margarida Gomes A et al. (2014) Adult B-cell acute lymphoblastic leukemia cells display decreased PTEN activity and constitutive hyperactivation of PI3K/Akt pathway despite high PTEN protein levels. Haematologica 99, 1062–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Borgo C et al. (2021) Protein kinase CK2: a potential therapeutic target for diverse human diseases. Signal Transduct. Target. Ther 6, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhou HX (2012) Intrinsic disorder: Signaling via highly specific but short-lived association. Trends Biochem. Sci 37, 43–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pedersen CP et al. (2020) Production of intrinsically disordered proteins for biophysical studies: Tips and tricks. Methods Mol. Biol 2141, 195–209 [DOI] [PubMed] [Google Scholar]
- 151.Dittmar G et al. (2019) PRISMA: Protein Interaction Screen on Peptide Matrix Reveals Interaction Footprints and Modifications- Dependent Interactome of Intrinsically Disordered C/EBPβ. iScience 13, 351–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Davey NE et al. (2017) Discovery of short linear motif-mediated interactions through phage display of intrinsically disordered regions of the human proteome. FEBS J. 284, 485–498 [DOI] [PubMed] [Google Scholar]
- 153.Krystkowiak I and Davey NE (2017) SLiMSearch: A framework for proteome-wide discovery and annotation of functional modules in intrinsically disordered regions. Nucleic Acids Res. 45, W464–W469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Palopoli N et al. (2015) QSLiM Finder: Improved short linear motif prediction using specific query protein data. Bioinformatics 31, 2284–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Prytuliak R et al. (2017) HH-MOTiF: De novo detection of short linear motifs in proteins by Hidden Markov Model comparisons. Nucleic Acids Res. 45, W470–W477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Martín M et al. (2022) MotSASi: Functional short linear motifs (SLiMs) prediction based on genomic single nucleotide variants and structural data. Biochimie 197, 59–73 [DOI] [PubMed] [Google Scholar]
- 157.Mészáros B et al. (2021) Mutations of intrinsically disordered protein regions can drive cancer but lack therapeutic strategies. Biomolecules 11, 1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mészáros B et al. (2021) Short linear motif candidates in the cell entry system used by SARS-CoV-2 and their potential therapeutic implications. Sci. Signal 14, eabd0334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Meyer K et al. (2018) Mutations in Disordered Regions Can Cause Disease by Creating Dileucine Motifs. Cell 175, 239–253.e17 [DOI] [PubMed] [Google Scholar]
- 160.Nishimura K et al. (2009) An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 6, 917–922 [DOI] [PubMed] [Google Scholar]
- 161.El Ashkar S et al. (2014) BET-independent MLV-based vectors target away from promoters and regulatory elements. Mol. Ther. - Nucleic Acids 3, e179–11 [DOI] [PMC free article] [PubMed] [Google Scholar]