Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo
- ️Fri Oct 12 2007
. Author manuscript; available in PMC: 2010 Dec 11.
Summary
Platelets play a central role in thrombosis, hemostasis, and inflammation. We show that activated platelets release inorganic polyphosphate (polyP), a polymer of 60-100 phosphate residues that directly bound to and activated the plasma protease factor XII. PolyP-driven factor XII-activation triggered release of the inflammatory mediator bradykinin by plasma kallikrein-mediated kininogen processing. PolyP increased vascular permeability and induced fluid extravasation in skin microvessels of mice. Mice deficient in factor XII or bradykinin receptors were resistant to polyP-induced leakage. PolyP initiated clotting of plasma via the contact pathway. Ablation of intrinsic coagulation pathway proteases factor XII and factor XI protected mice from polyP-triggered lethal pulmonary embolism. Targeting polyP with phosphatases interfered with procoagulant activity of activated platelets and blocked platelet-induced thrombosis in mice. Infusion of polyP restored defective plasma clotting of Hermansky-Pudlak Syndrome patients, which lack platelet polyP. The data identify polyP as a new class of mediator having fundamental roles in platelet-driven proinflammatory and procoagulant disorders.
Introduction
In mammals, blood clotting involves both cellular and protein components, provided by platelets and coagulation factors, respectively. Upon vascular injury, platelets rapidly adhere and aggregate to form a plug; simultaneously, coagulation factors respond in a complex reaction cascade to form fibrin, which strengthens platelet clots (Furie and Furie, 2008; Mackman, 2008). It has become evident that platelets also contribute to host responses to infection and inflammatory vascular processes such as formation and extension of atherosclerotic plaques (Ruggeri, 2002). Basic and clinical data suggest that thrombosis and inflammation are intrinsically linked and share several key mechanisms (von Hundelshausen and Weber, 2007; Wagner and Frenette, 2008).
In the original cascade/waterfall model of coagulation, thrombin and fibrin formation is initiated by two distinct pathways, triggered by exposure of blood to a damaged vessel wall (extrinsic) or to blood-borne (intrinsic) factors. The intrinsic pathway of coagulation is initiated by factor XII (FXII), in a reaction involving high molecular weight kininogen (HK) and plasma kallikrein (PK), collectively referred to as the plasma contact system. Upon contact with anionic surfaces a conformational change occurs in FXII zymogen, resulting in a small amount of active FXII (FXIIa). FXIIa cleaves PK to generate active kallikrein, which in turn reciprocally activates additional FXII (Colman, 2006). This process triggers the intrinsic pathway of coagulation via FXIIa-mediated activation of factor XI (FXI), and also liberates the inflammatory mediator bradykinin (BK) by kallikrein-mediated cleavage of HK (Gailani and Renne, 2007a). The binding of BK to its B2 receptor (B2R) activates various intracellular signaling pathways that dilate vessels, induce neutrophil chemotaxis, and increase vascular permeability (Leeb-Lundberg et al., 2005).
FXII is activated in vitro by a variety of polyanionic surfaces, such as kaolin, glass, ellagic acid, certain polymers, nucleotides, sulfatides, misfolded proteins, and some types of collagen or glycosaminoglycans (Muller and Renne, 2008). FXII activation by kaolin (a silicate) is used to trigger the aPTT clotting assay that is a standard laboratory measurement of plasma coagulation. Despite its importance for in vitro clotting, FXII deficiency is not associated with any hemorrhagic disorder in humans or mice (Ratnoff and Colopy, 1955; Pauer et al., 2004) and is considered to be redundant for normal hemostasis. Challenging the dogma of the coagulation balance, mouse models indicate a critical role of FXII in thrombosis. Deficiency of FXII attenuates arterial thrombus formation and protects animals from ischemic brain injury (Renne et al., 2005a; Kleinschnitz et al., 2006). For >40 years platelets have been linked to the intrinsic pathway (Castaldi et al., 1965), with activation promoting fibrin formation in a FXII-dependent manner (Walsh and Griffin, 1981). These findings suggest that FXII is activated on procoagulant platelets but the mechanisms involved and the relevance of platelet-driven procoagulant and proinflammatory reactions in vivo are unclear (Furie and Furie, 2008).
Polyphosphate (polyP) is an inorganic, linear polymer of orthophosphate units linked by phosphoanhydride bonds. PolyP is abundant in nature and has been conserved throughout evolution. PolyP has been extensively studied in prokaryotes and lower eukaryotes, where it functions in basic metabolism, and stress responses and as a structural component (Rao et al., 2009). Dense granules of platelets contain polyP (Ruiz et al., 2004). Synthetic polyP is a potent modulator of plasma clotting, affecting the intrinsic pathway, the fibrinolytic system, factor V activation, and fibrin structure (Smith and Morrissey, 2008a; Smith et al., 2006). The in vivo relevance of polyP functions remains to be established.
Using genetically altered mice and patient plasma we show that platelet polyP is a procoagulant and proinflammatory mediator by activating the plasma contact activation system. PolyP represents the long sought “foreign” surface that triggers fibrin formation by activated platelets linking primary to secondary hemostasis and critically contributing to “procoagulant” platelet activity. Interrupting polyP-driven fibrin- and BK-generation interferes with thrombosis and edema formation in vivo may present a new paradigm to fight thromboembolic and inflammatory disease.
Results
Activated platelets release polyP
Platelet dense granules contain polyP (Ruiz et al., 2004). We analyzed whether activated/procoagulant platelets release polyP. Washed human platelets were stimulated with ADP, thrombin receptor-activating peptide 6 (Trap6), collagen, or thrombin, after which polyP was isolated from stimulated platelet supernatants using an ion-exchange method previously used to purify polyP from cell lysates (Kumble and Kornberg, 1995). It was then separated by agarose-gel electrophoresis and probed with toluidine blue (Figure 1A) and a more sensitive DAPI-based fluorescent stain (Figure 1B) (Smith and Morrissey, 2007). Incubation of the extracted material with proteinase, RNase, DNase, or glycosaminoglycan-cleaving enzymes did not alter the staining patterns (not shown). In contrast, treatment with phosphatase (Psp), which hydrolyzes polyP, completely abolished the signal (Figures 1A and B, “thrombin+Psp”). Platelet polyP migrated in a similar manner to synthetic polyP with mean chain lengths of 70 ± 16 (polyP70) phosphate units and at higher mass than polymers of 20 ± 6 phosphates (polyP20). We determined the chain length of platelet polyP using 31P-NMR. The relative ratio of signal intensities of the phosphate end groups (α-P, -10.2 ppm) versus central phosphates (β-P, -22.1 ppm) yielded a mean chain length of platelet polyP of 80, consistent with its migration in the agarose matrix (Figure 1C). Our NMR analysis indicated that the mean chain length of “polyP75” used in previous studies (Ruiz et al., 2004; Smith et al., 2006) is 125 (Figure S1). This polyP (termed polyP125, Figures 1A and B) migrated with higher molecular weight than platelet polyP. Integration of all 31P signals revealed that >69 % of total platelet released inorganic phosphate is polyP.
Figure 1. Activated platelets secrete long-chain polyP.

Washed platelets (3.8×1013 platelets each) were stimulated with ADP (0.1 mM), Trap6 (0.2 mM), collagen (0.2 mg/ml), thrombin (2 U/ml), or buffer (“w/o”) and polyP was isolated from the supernatants. PolyP were separated by agarose gel electrophoresis and stained with toluidine blue (A) or DAPI (B). Synthetic polyP with mean chain lengths of 20 (polyP20), 70 (polyP70) or 125 (polyP125) phosphate units were loaded in lanes 1-3 as size standards. Purified material from thrombin-stimulated platelets was incubated with Psp (0.05 U/μg polyP; “thrombin + Psp”) prior to electrophoresis. Negatively charged chondroitin sulfate (CS) confirmed specificity of staining. A DNA ladder as size standard is indicated on the left. (C) 31P NMR spectrum of platelet polyP at 202.4 MHz. The terminal and internal phosphates of the polyP chain give signals at 10.2 (doublet, blue arrow) and 22.1 (red arrow) ppm, respectively; ratios of their intensities yielded a mean chain length of 80 for platelet polyP. Other phosphate species present are monophosphate (0.0 ppm, 5 % of total platelet released phosphate), diphosphate (-10.4 ppm, 13 %), cyclotriphosphate (-21.4 ppm, 10 %) and ADP (-11.2 ppm, 3 %).
PolyP activates the contact system in vitro
We analyzed whether platelet polyP could act as a surface for FXII activation using the chromogenic substrate S-2302, which is hydrolyzed by active FXII (FXIIa) (Figure 2A). Addition of polyP (100 μg/ml) to human (or to murine, Figure S2A) platelet-free plasma (PFP) generated FXIIa with similar kinetics and potency as an identical concentration of kaolin. Treatment of polyP with Psp before addition to plasma reduced polyP-driven FXIIa generation in human and murine plasma in a time-dependent manner. Psp digestion for >60 min completely abolished the FXIIa-generating activity of polyP (Figures 2A and S2A), suggesting that a critical polymer length is required to induce FXII activation. Indeed, synthetic polyP of mean chain length ≤45 phosphate units failed to trigger FXII activation in plasma (Figure S2B). To discriminate between polyP-mediated FXII autoactivation and FXII activation by PK, we incubated purified FXII and PK with polyP. PolyP was as efficient as dextran sulfate (a strong contact activator; Colman, 2006) in supporting PK-driven activation of FXII (Figure 2B). When autoactivation of FXII was investigated without the addition of PK to the reaction, polyP generated significant amounts of FXIIa compared to the no surface control but was not as efficient as dextran sulfate (Figure 2C). The ability of a molecule to trigger contact activation relies on interactions of contact factors with the negatively charged surface (Colman, 2006). A gel shift assay demonstrated direct binding of polyP to contact system factors (Figure 2D). Electrophoretic mobility shifts indicated strong interaction of polyP with both FXII and HK, whereas binding to PK and FXI was minor. PolyP did not bind to antithrombin or albumin. Zirconia beads coated with either polyP or albumin confirmed specific and direct polyP binding to FXII, PK, HK, and FXI, with FXII being the strongest polyP-interacting protein (Figure 2E).
Figure 2. PolyP triggers contact activation of factor XII.

(A) Human plasma was incubated with 100 μg/ml polyP (▲) or kaolin (○). Platelet polyP was either untreated (0) or pretreated with Psp (0.05 U/μg polyP) for 10 ( ), 30 (
), 30 ( ), or 60 min (
), or 60 min ( ). Data are means ± SD, n=5. (B and C) Plasma-free activation of purified FXII (200 nM) was analyzed in the presence (B) or absence (C) of PK (0.5 nM). The mixture was stimulated with dextran sulfate (■; 1 μg/ml), polyP (▲; 5 μg/ml), or buffer (●). FXIIa concentrations were determined using S-2302 hydrolysis. Data are means ± SD, n=4. (D) Gel-mobility shift analyses of polyP binding to proteins. Purified FXI, FXII, PK, HK, antithrombin (AT) or albumin (ALB) were incubated with buffer (“w/o”) or with increasing concentrations of polyP125 (0 - 100 μg) for 10 min, after which mixtures were resolved on a 10 % Tris-Glycine gel under native conditions and stained for protein using Gel Code Blue. (E) Protein binding to immobilized polyP. PolyP (left) or albumin (ALB, right) was coupled to zirconia (zirconium dioxide) beads. FXII, FXI, PK, or HK (5 μg each) were incubated with the beads, after which the flow-through (“FT”) fraction was collected by centrifugation using mini spin columns. The beads were washed with a low salt buffer (50 mM NaCl; “LS”) followed by a high salt buffer (1 M NaCl; “HS”). Fractions were probed by Western blotting with appropriate antibodies. Starting material (“SM”) refers to the initial protein load added to the beads.
). Data are means ± SD, n=5. (B and C) Plasma-free activation of purified FXII (200 nM) was analyzed in the presence (B) or absence (C) of PK (0.5 nM). The mixture was stimulated with dextran sulfate (■; 1 μg/ml), polyP (▲; 5 μg/ml), or buffer (●). FXIIa concentrations were determined using S-2302 hydrolysis. Data are means ± SD, n=4. (D) Gel-mobility shift analyses of polyP binding to proteins. Purified FXI, FXII, PK, HK, antithrombin (AT) or albumin (ALB) were incubated with buffer (“w/o”) or with increasing concentrations of polyP125 (0 - 100 μg) for 10 min, after which mixtures were resolved on a 10 % Tris-Glycine gel under native conditions and stained for protein using Gel Code Blue. (E) Protein binding to immobilized polyP. PolyP (left) or albumin (ALB, right) was coupled to zirconia (zirconium dioxide) beads. FXII, FXI, PK, or HK (5 μg each) were incubated with the beads, after which the flow-through (“FT”) fraction was collected by centrifugation using mini spin columns. The beads were washed with a low salt buffer (50 mM NaCl; “LS”) followed by a high salt buffer (1 M NaCl; “HS”). Fractions were probed by Western blotting with appropriate antibodies. Starting material (“SM”) refers to the initial protein load added to the beads.
PolyP triggers bradykinin generation
We examined whether polyP stimulate BK release in human plasma and observed complete cleavage of HK at concentrations >1 μg/ml (Figure 3A). Consistently, BK concentration in these samples was high (>750 ng/ml), but relatively low in samples treated with buffer (31 ± 4 ng/ml) or ≤1 μg/ml polyP, which was not sufficient to initiate HK processing (Figure 3B). To confirm that the FXII/PK cascade mediated BK generation from HK, we probed for the zymogens by Western blotting and found that polyP >1 μg/ml triggered complete PK and FXII activation with a slightly higher concentration (>2 μg/ml) resulting in complete activation of plasma FXII (Figure 3A).
Figure 3. PolyP activates the kallikrein-kinin system.

(A) PolyP-induced cleavage of FXII, PK and HK in plasma. Human plasma was incubated with increasing concentrations of polyP (0.5 - 1000 μg/ml) or buffer (“w/o”), and then subjected to SDS-PAGE and Western blotting. Samples were probed with antibodies against FXII, HK, and PK. (B) PolyP-induced bradykinin release in plasma. BK in polyP-incubated samples was quantified by ELISA. Data are means ± SD, n=4.
PolyP initiates bradykinin-dependent mechanism in vivo
To analyze whether polyP initiates contact system-mediated capillary leakage via BK formation, we employed a Miles edema model in genetically altered mice (Figures 4A-F). In WT animals, platelet polyP or synthetic polyP125 (10 μg each) initiated leakage was increased 7.9 ± 0.8 and 7.4 ± 1.0 fold, respectively, higher than vehicle alone (Figures 4A and G). BK stimulation in WT mice increased leakage to 11.3 ± 1.5 fold over vehicle (Figure 4A). To confirm that polyP induces vascular leakage by releasing BK, we employed B2R-deficient (B2R-/-) mice, which are protected from BK-driven edema formation (Han et al., 2002). B2R-/-mice were mostly resistant to polyP-induced increase in permeability (polyP 1.1 ± 0.2 fold and polyP125 1.1 ± 0.1 fold, Figures 4B and G). Similarly, BK failed to induce a response in B2R-/- mice (1.1 ± 0.2 fold, not depicted) whereas injection of histamine (100 μM) resulted in considerable leakage (7.7 ± 1.3 fold compared to vehicle), confirming that B2R-/- mice are susceptible to edema formation via contact system independent pathways. Because FXII-independent mechanisms for PK activation exist (Muller and Renne, 2008), we analyzed polyP-triggered leakage in FXII-deficient mice (FXII-/-), which are defective in contact system-driven BK formation (Pauer et al., 2004). FXII-/- mice were almost completely resistant to polyP-induced leakage, and the degree of edema was not significantly different from the buffer-treated group (polyP 1.3 ± 0.2 fold and polyP125 1.2 ± 0.2 fold, n=10, p>0.05, Figures 4C and G). BK-stimulated edema formation in FXII-/- mice was similar to that observed in WT animals (10.4 ± 1.3 fold).
Figure 4. PolyP triggers contact system-mediated bradykinin-induced edema in mice.

(A-F) Evans blue was intravenously infused as a tracer into the following: (A) WT mice, (B) B2R-/-mice, (C) FXII-/-mice, (D) C1INH-/- mice, (E) PCK-infused WT mice (8 mg/kg body weight), and (F) Icat-administered WT mice (175 μg/kg body weight). Dorsal skin edema formation was induced by intradermal injection of 50 μl normal saline (NaCl), BK (100 μM), platelet polyP (10 μg), or synthetic polyP125 (10 μg), respectively, and visualized by tracer extravasation after 30 min. (G) Evans blue tracer from skin welts was extracted and quantified. Extravasated tracer is plotted as fold-increase of saline-induced signal in WT mice, to control for inter-animal variability. Data are means ± SD, n=10. (H) Psp- (0.05 U/μg polyP, 30 min; “Psp”) or buffer-treated E. coli polyP (“w/o”, 2 μg/lane each) were separated by agarose gel electrophoresis and stained with toluidine blue. A DNA ladder as size standard is indicated on the left. (I, J) Murine plasma was incubated with Psp (“Psp”) or buffer-treated (“w/o”) E. coli polyP (1 μg/ml, 30 min) and was probed for FXII, FXI, PK zymogen forms, single-chain HK, and LK by Western blotting. ELISA determined BK in samples supplemented with polyP. Means ± SD, n=5. (K) Mortality associated with intraperitoneal injection of E. coli polyP (750 μg/g body weight) in WT, FXII-/-, or B2R-/- mice. For control, polyP was Psp digested prior to infusion into WT animals (“+Psp”). Animals alive in each group (n=10) 30 min after challenge, were considered as survivors.
Hereditary angioedema is characterized by recurrent attacks of swelling owing to a deficiency of a functional C1 esterase inhibitor (C1INH), which is the major plasma inhibitor for several complement proteases, FXIIa and PK (Zuraw, 2008). PolyP initiated edema in C1INH null (C1INH-/-) animals that exceeded levels in WT by >40% (polyP 11.8 ± 0.9 fold and polyP125 10.9 ± 0.8 fold, Figures 4D and G), whereas BK-induced (11.0 ± 0.8 fold) tracer extravasation was similar to WT mice levels (11.3 ± 1.5 fold).
Since congenital deficiency in B2R or FXII protects mice from polyP-driven edema formation, pharmacological targeting of BK-signaling or -formation should provide similar protection. To address this hypothesis, we treated WT mice with the FXIIa inhibitor H-D-Pro-Phe-Arg-chloromethylketone (PCK) (Kleinschnitz et al., 2006), or with the B2R antagonist icatibant (Icat), 5 min prior to polyP application. PolyP was unable to induce leakage in WT mice pretreated with PCK (polyP 1.9 ± 0.4 fold and polyP125 1.2 ± 0.2 fold, Figures 4E and G) or Icat (polyP 1.6 ± 0.2 fold, polyP125 1.1 ± 0.2 fold, Figures 4F and G). BK function was blocked by Icat (1.2 ± 0.1 fold) but was independent of FXII activity (9.6 ± 2.3 fold, Figure 4F).
BK is an important mediator in bacterial infections. Escherichia coli that locally assemble contact system proteins on their surface generate BK (Herwald et al., 1998). We analyzed bacterial polyP for BK formation. PolyP was purified from E. coli, separated by agarose-gel electrophoresis, and stained with toluidine blue. Consistent with chain lengths of 100 - 1000 in E. coli (Rao et al., 2009) the polymer migrated with an apparent molecular weight of 10 -100 kDa (Figure 4H). As little as 1 μg/ml E. coli-derived polyP initiated contact activation with complete cleavage of FXII, FXI, PK, and HK, but not of low molecular weight kininogen (LK) (Figure 4I); E. coli polyP potently triggered BK generation (Figure 4J). Psp treatment of E.coli polyP abrogated activation of the contact system, as detected by persistence of zymogen forms (right lanes, Figure 4I) and low levels of BK (28 ± 3 ng/ml, Figure 4J). Intraperitoneal application of polyP (750 μg/g body weight) in WT mice induced strong writhing reactions (a pain reaction) and decreased systemic arterial blood pressure (109 ± 17 to 57 ± 39 mmHg, n=10, survivors and non-survivors); 9/10 mice died within 15 min of challenge (Figure 4K). In contrast, FXII-/- and B2R-/- mice were protected from polyP-induced hypotonic shock, writhing reaction, edema formation, with 8/10 and 7/10 animals (**p<0.01 FXII-/- or B2R-/- vs. WT), respectively, surviving.
PolyP initiates the intrinsic pathway of coagulation in plasma
To analyze the procoagulant activity of polyP, we incubated human plasma with increasing concentrations of the polymer (0.5 - 1000 μg/ml) and followed generation of FXIIa and activation of its principal substrate in the intrinsic pathway of coagulation, FXI, by Western blotting (Figure 5A); we also quantified thrombin generation (Figure 5B). PolyP ≥2 μg/ml triggered the conversion of FXII and FXI to active proteases (as indicated by the disappearance of the zymogen forms at 80 kDa, Figure 5A), which correlated with polyP-triggered thrombin generation (Figure 5B). Digestion of polyP with Psp (0.05 U/μg polyP) impeded the procoagulant activity of the polymer. Furthermore, we compared polyP with kaolin for initiating thrombin formation in plasma in real time using a fluorogenic thrombin substrate. PolyP- and kaolin-driven thrombin generation had almost identical kinetics, potency, and effects on total thrombin generation (Figure S2C). Pre-incubation of polyP with Psp largely abolished thrombin formation (ETP <100 nM*min). Addition of polyP to normal plasma but not FXII-depleted plasma shortened the clotting time in a dose-dependent manner (Figure 5C). Clotting times in recalcified plasma triggered by synthetic polyP125 and polyP1000 were similar to kaolin, whereas polyP3 stimulated clotting was similar to buffer control (Figure S2D).
Figure 5. PolyP activates the intrinsic pathway of coagulation in plasma.
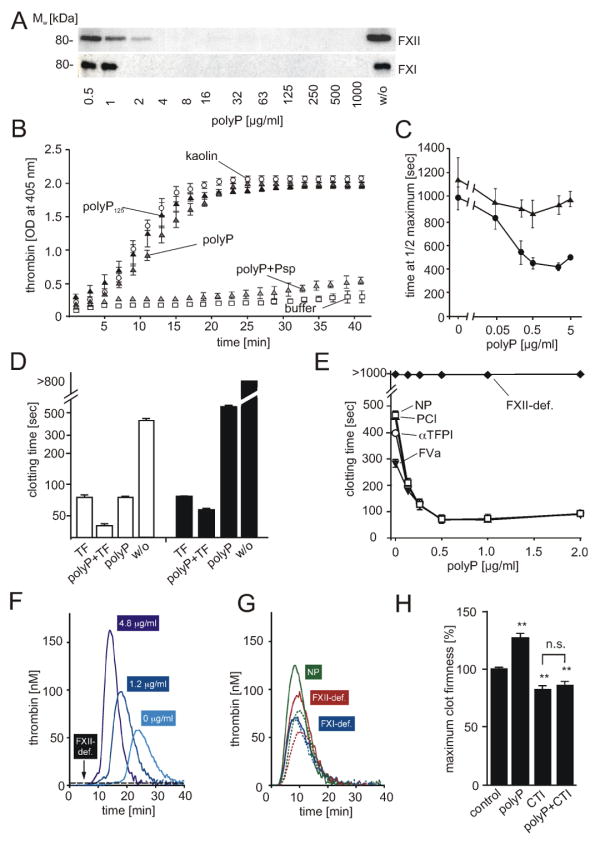
(A) PolyP-induced cleavage of FXII and FXI in plasma. Plasma was incubated for 30 min at 37°C with increasing concentrations of polyP (0.5 - 1000 μg/ml) and analyzed for zymogen forms of FXII and FXI by Western blotting. Untreated plasma (“w/o”) shows initial FXII and FXI levels. (B) Thrombin generation in plasma incubated with buffer alone (□), 100 μg/ml kaolin (○), or platelet polyP (; “polyP”), synthetic polyP125 (▲), platelet polyP treated with Psp prior to addition of plasma (; “polyP+Psp”). Data are means ± SD, n=5. (C) Plasma clotting times. PolyP was added to normal plasma (●) or FXII-immunodepleted plasma (▲) at a range of concentrations (0 - 5 μg/ml), with clot formation quantified as the half-maximal change in turbidity at 405 nm. Data are means ± SD, n=3. (D) Recalcification clotting times in normal (white columns) and FXII-deficient human plasma (black columns) incubated with TF (1 pM), polyP (3 μg/ml), TF and polyP (1 pM and 3 μg/ml), or buffer (w/o). Data are means ± SD, n=5. (E) Recalcification clotting times triggered by polyP (0 - 2 μg/ml) in FXII-deficient plasma (FXII-def.) and normal human plasma (NP) supplemented with FVa (4 nM, ▼), αTFPI antibody (10 μg/ml, □), or PCI (30 μg/ml, ○). Means ± SD, n=3. (F-H) Real time thrombin generation: (F) PolyP (0; 1.2; 4.8 μg/ml) triggered thrombin formation in plasma preincubated with an extrinsic pathway-inhibitor FVIIai. PolyP (4.8 μg/ml) failed to generate thrombin in FVIIai treated FXII-deficient plasma (FXII-def.). (G) Thrombin generation triggered by TF in FXII-deficient (red), FXI-deficient (blue), and normal plasma (green) in the absence (dashed lines) or presence of polyP (solid lines, 4.8 μg/ml). (H) Maximum clot firmness assessed by thromboelastography: Clot firmness in polyP (5 μg/ml), CTI (100 μg/ml), or polyP plus CTI-treated whole blood relative to untreated samples (100 %). Data are means ± SD, n=5.
We analyzed tissue factor (TF)- and polyP-initiated clotting in normal and FXII deficient human plasma (Figure 5D). Application of TF or polyP shortened the clotting time to 80 ± 6 and 78 ± 5 sec, respectively (untreated plasma >450 sec). PolyP addition to TF-stimulated plasma further accelerated clotting (29 ± 5 sec). The ability of polyP to induce clotting in FXII deficient plasma was impaired (590 ± 35 sec) compared to normal plasma, whereas TF-stimulated clotting was unaffected (82 ± 2 sec). Co-application of polyP and TF to FXII-deficient plasma further shortened clotting times slightly to 66 ± 3 sec. Similar to human plasma, polyP-induced clotting of WT mouse plasma (74 ± 5 sec) and accelerated TF-initiated clotting (TF alone: 69 ± 5, TF and polyP: 54 ± 6 sec). In FXII-/- mouse plasma, polyP had minor procoagulant activity (380 ± 42 sec) and slightly increased TF-initiated clotting (76 ± 5 vs. 65 ± 4 sec) (Figure S3A).
PolyP also acts on proteins downstream of FXII in the coagulation cascade such as tissue factor pathway inhibitor (TFPI), factor V (FV), and thrombin-activatable fibrinolysis inhibitor (TAFI) (Smith et al., 2006). We analyzed the relative importance of coagulation initiation via FXII versus acceleration of fibrin formation via FV, TFPI, and TAFI for procoagulant polyP-activity (Figure 5E). Plasma was incubated with increasing polyP concentrations (0 - 2 μg/ml, not sufficient to completely activate FXII) in the presence or absence of active FV (FVa), PCI (a TAFI inhibitor), or TFPI function-inhibiting antibodies. Adding exogenous FVa to plasma accelerated clotting somewhat in the absence of polyP. The procoagulant activity initiated by as little as 0.5 μg/ml polyP, however, greatly exceeded polyP-effects on FV (Figure 5E). PolyP-driven clotting was largely independent of anti-TFPI antibodies or PCI in this assay. PolyP completely failed to trigger clotting in FXII deficient plasma, indicating that polyP effects on downstream coagulation proteins are not sufficient to induce clotting without FXII activation. To specifically analyze polyP-stimulated thrombin generation via the intrinsic pathway, we blocked TF/FVII activity with 30 pM FVIIai. In a dose dependent fashion, polyP (0 - 4.8 μg/ml) increased total and maximal thrombin (ETP, 569-971 nM*min, peak: 58-163 nM), and shortened lag time (19.5-10.5 min) and time to peak (24-14 min) (Figure 5F). In the absence of TF/FVII activity there was no effect of polyP on thrombin generation in FXII deficient plasma (FXII-def., Figure 5F). In contrast, polyP accelerated thrombin generation in FXII-deficient plasma stimulated with TF (0.5 pM), although not by a large amount. PolyP did not increase thrombin formation stimulated by TF in FXI-deficient plasma (Figure 5G). Neither inhibition of TAFI or TFPI activity affected polyP–driven thrombin formation in the presence or absence of TF/FVII activity (not depicted), arguing against direct effects of the polymer on these proteins.
The originally described patient with FXII-deficiency, John Hageman, died from pulmonary embolism, which apparently originated from a hemipelvis fracture-associated deep vein thrombosis (Ratnoff et al., 1968). Does platelet polyP-initiated FXII activation have implications for the embolic disease of Mr. Hageman? We used thromboelastography in whole blood to analyze clot firmness dependent on polyP and FXII activity (Figure 5H). This method measures mechanical clot stability (represented by the maximal amplitude of the curves in Figure S3B). Addition of polyP (5 μg/ml) increased clot firmness to 128 ± 4 % vs. untreated blood (100%). Pharmacological targeting of FXII activity abolished polyP-mediated clot stabilization (85 ± 3 vs. 83 ± 3 % in CTI treated blood, p>0.05). As polyP does not affect factor XIII (FXIII) activity or activation (Figure S3C), increased clot firmness conferred by the polymer appears to be mediated by enhanced fibrin structures (Smith and Morrissey, 2008a) and fibrin production (Figure 5F).
Deficiencies in intrinsic pathway proteases protect mice from polyP-induced thrombosis
To analyze polyP for fibrin formation in vivo, we challenged WT and FXII-/- mice in a model of lethal pulmonary thromboembolism (PE) by intravenous infusion of polyP (300 μg/g body weight). All WT mice with the exception of a single animal (14/15) died within 5 min of polyP application (Figure 6A). In contrast, FXII-/- mice were significantly protected from polyP-induced PE, with 12 out of 15 FXII-/- mice surviving the challenge for >30 min (**p<0.01 FXII-/- vs. WT). To show that the high level of survivors observed in FXII-/- mice was a direct result of defective polyP-driven FXII activation, we reconstituted FXII null mice with human FXII (hFXII). Intravenous infusion of human protein corrected the prolonged aPTT clotting time of FXII-deficient murine plasma to normal values of WT mice (28 ± 4 sec) and restored susceptibility to lethal PE after polyP infusion, with 12 out of 15 reconstituted animals dying (Figure 6A). Pretreatment of polyP with Psp abolished its procoagulant activity, with 14/15 mice surviving infusion of degraded polyP (Figure 6B). The infestin4-based recombinant FXIIa-inhibitor CSL829 (Schmidtbauer et al., 2009) and PCK irreversibly inhibit the amidolytic activity of FXIIa and PK-mediated activation of FXII. CSL829 blocked polyP-driven FXII and consecutive FXI, PK, and HK activation steps in plasma (Figure 6C). To test the protective potential of FXIIa-inhibitors for polyP-driven thrombosis, WT mice were intravenously injected with CSL829 10 min prior to polyP infusion, which prolonged the aPTT (48 ± 9 sec) and significantly protected mice from lethal PE (12 out of 15 survived; **p<0.01 vs. WT, Figure 6A). PCK (8 μg/g body weight) provided similar protection from polyP-triggered thromboembolism (12/15 survived, not shown). Histological sections of lung tissue from polyP treated mice and counting of formed thrombi are shown in Figures 6D and E. While the majority of vessels was obstructed in WT and hFXII-reconstituted FXII-/- mice (dead and survivors), virtually no thrombi were found in FXII-/- mice and in WT animals treated with FXIIa-inhibitors.
Figure 6. PolyP triggers thrombosis in vivo.

(A and B) Survival times following polyP challenge. (A) Pulmonary thromboembolism was induced by i. v. infusion of platelet polyP (300 μg/g body weight) in WT mice, FXII-/- mice, FXII-/- mice reconstituted with human FXII (“hFXII”, 2 μg/g body weight), WT mice infused with FXIIa inhibitor CSL829 (15 μg/g body weight), FXI-/- mice, FXII- and FXI-gene double-deficient (“FXII-/-/FXI-/-”) mice, or B2R-/- mice. Mortality was assessed in each group (n=15); animals still alive 30 min after challenge, were considered survivors. (B) PolyP was Psp digested (0.05 U/μg polyP) prior to infusion into WT animals and survival was analyzed as in panel A. (C) Plasma supplemented with CSL829 (150 μg/ml) or buffer (“w/o”) was incubated with polyP (500 μg/ml) for 30 min and analyzed for zymogen FXII, FXI, PK and HK by Western blotting. (D) Hematoxylin and eosin stained sections of lungs of WT, FXII-/-, CSL829 infused WT, and hFXII reconstituted FXII-/- mice 30 min after polyP administration (bar = 100 μm). (E) Thrombi per visual field were counted at 10× magnification from sections such as those in panel D. Data are mean ± SD for 100 fields. (F) Accumulation of fibrin in lung tissue of polyP treated WT, FXII-/-, CSL829 pretreated WT, and reconstituted FXII-/- mice. Fibrin formation 30 min after polyP challenge was analyzed by Western blotting.
To analyze the mechanism of polyP-initiated PE in vivo we used FXI-/- mice and generated FXII-/-/FXI-/--double gene deficient animals. If the prothrombotic effect of polyP is mediated through FXII activation and the consecutive activation of FXI by FXIIa, then FXI-/- and FXII-/-/FXI-/--deficient animals should be protected from PE to the same extent as FXII-/- mice (Figure 6A). Indeed, FXI and FXII/FXI-combined deficiency conferred resistance to polyP-induced PE and both mouse strains were largely protected from polyP challenge (11/15 and 13/15 survived; **p<0.01 FXI-/- and FXII-/-/FXI-/- vs. WT; p>0.05 FXI-/- and FXII-/-/FXI-/- vs. FXII-/-). Targeting B2R was shown to confer protection from arterial occlusion induced by vascular injury (Gailani and Renne, 2007a). We analyzed B2R-/- mice in our PE model and found that almost all (13/15) animals died, suggesting that BK is not required for polyP initiated PE. To confirm that polyP-initiated FXII activity contributes to pathological thrombosis by the intrinsic pathway of coagulation, we measured fibrin formation in lung tissues by quantitative immunoblot analysis of urea-insoluble tissue extracts with the fibrin specific antibody 59D8. Fibrin accumulation was significantly reduced in the lungs of FXII-/- and inhibitor treated mice, compared to WT and reconstituted animals (Figure 6F).
PolyP initiates fibrin formation on activated platelets
To assess whether polyP may present the “foreign surface” that initiates FXII-driven fibrin formation on platelets we determined whether Psp, which efficiently degrades polyP (Figure 2A), inhibits procoagulant platelet activity. Platelets of human (Figure 7A) and mouse (Figure 7B) origin were stimulated with calcium ionophore A23187 prior to recalcification. Platelet activation reduced clotting times in both species up to 3.2- and 4.2-fold compared to recalcification times in unstimulated PRP. Addition of Psp prior to recalcification abrogated the procoagulant activity of stimulated platelets resulting in similar clotting times to unstimulated PRP. Psp did not alter the recalcification time in PRP without platelet activation. Consistent with our initial observation that clotting in A23187-stimulated PRP depends on FXII (Renne et al., 2005a), clotting in FXII deficient PRP in response to the platelet activator was severely impaired in the presence or absence of Psp (Figures S4A and S4B).
Figure 7. Targeting polyP blocks platelet procoagulant activity in vitro and thrombosis in vivo.

(A and B) Recalcification clotting times were determined in platelet-rich (A) human or (B) WT mouse plasma stimulated with A23187 (5 μM) in the presence (+) or absence (-) of Psp (10 U/ml). Reductions in clotting times are given relative to untreated plasma. Data are means ± SD, n=6. (C) Recalcification clotting times were determined in Trap6 (30 μM) stimulated human platelet-rich plasma in the presence (+) or absence (-) of PGE1 (5 μM), dBcAMP (0.5 mM), and Psp (10 U/ml). Reductions in clotting times are given relative to untreated PRP. Data are means ± SD, n=6. (D) Mortality associated with i.v. injection of 0.7 μg/g body weight Trap6 in WT mice; WT mice i.v. infused with PGE1 (0.85 μg/g body weight “WT+PGE1”) or dBcAMP (100 μg/g body weight “WT+dBcAMP”); FXII-/- mice; and WT mice injected i.v. with Psp (15 U/g body weight; “WT+Psp”) before Trap6 challenge. 13 of 15 WT mice died within 10 min of challenge. In contrast WT animals injected with PGE1 or dBcAMP, FXII-/-, and Psp-treated WT mice were significantly protected from lethal pulmonary embolism induced by Trap6; 10/15, 10/15, 13/15, and 13/15, respectively, survived for >30 min each (WT+PGE1, WT+dBcAPMP, FXII-/- and WT+Psp vs. WT p<0.01, n=15 per genotype). (E) PolyP “rescues” defective fibrin formation in Hermansky-Pudlak Syndrome patients. Human platelets were isolated from healthy blood donors (control) and from patients with Hermansky-Pudlak Syndrome (HPS) and stimulated for 10 min at 37°C with Trap6 (50 μM) and collagen (10 μg/ml). Recalcification clotting times in normal plasma on addition of either normal (control) or HPS platelets (HPS, 1.5 ×107) in the absence (open bars) or presence (closed bars) of additional synthetic polyP125 (10 μg/ml). The graph shown is representative of 3 different experiments performed on different HPS and normal individuals.
We analyzed whether platelet inhibitors, prostaglandin E1 (PGE1) and N6,2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate (dBcAMP), which interfere with thrombin-stimulated platelet activation, could inhibit the procoagulant activity of Trap6-stimulated human (Figure 7C) and murine (Figure S4C) platelets. PGE1, dBcAMP or Psp did not alter recalcification times in PRP without platelet activation. Trap6 reduced clotting times 3.2-fold in human and 2.6-fold in mouse PRP. Addition of Psp prior to recalcification interfered with Trap6 enhanced clotting (1.2- in human and 1.4-fold in mouse PRP compared to unstimulated control). Application of PGE1 and dBcAMP prior to Trap6 reduced procoagulant platelet activity (1.9- and 1.6-fold in human and 1.6- and 1.8-fold in murine PRP). Treatment with Psp in addition to PGE1 or dBcAMP further reduced the procoagulant activity of Trap6-treated platelets until clotting times were no longer significantly different from unstimulated PRP (p>0.05).
To analyze polyP functions for pathological clotting driven by stimulated platelets in vivo, we established a model of lethal PE initiated by Trap6 infusion (Figure 7D). Trap6 activates platelets and initiates polyP secretion (Figure 1), but Trap6 by itself has no direct effect on the plasma coagulation cascade. Almost all (13/15) WT mice died within 5 min after intravenous infusion of Trap6 (Figure 7D). In contrast, FXII-/- mice were largely protected from Trap6-induced lethal PE (13/15 survived). Consistent with anticoagulant activity in human and mouse plasma (Figures 7C and S4C), PGE1 and dBcAMP infusion into WT mice conferred protection from PE; 10/15 mice treated with each inhibitor survived the Trap6 challenge. Infusion of Psp prior to Trap6 application protected WT mice from lethal PE and 13 out of 15 animals survived the challenge for >30 min. Lung histology from Trap6 treated mice confirmed PE (Figure S4D). While the vast majority of vessels was obstructed in WT animals, virtually no thrombi were found in FXII-/- mice and in WT mice treated with Psp. Endogenous Psp activity in plasma (Smith et al., 2006) and on endothelial cells (Figures S5A and S5B) degrades polyP slowly and polyP released from activated platelets coincubated with endothelial cells (Figure S5D, inset) initiated FXII-dependent thrombin and BK formation (Figures S5C-E).
We further tested the importance of this concept for human disease states. Hermansky-Pudlak Syndrome (HPS) is a rare, complex hereditary disease (Gahl et al., 1998), in which a bleeding diathesis results from platelet storage pool deficiency. HPS patients have reduced or absent platelet dense granules, the polyP storage site in platelets. We analyzed platelets isolated from HPS patients for their ability to initiate clotting (Figure 7E). Normal or HPS platelets were stimulated with Trap6 before adding them to normal PFP. Supplementing plasma with normal platelets resulted in a recalcification time of 280 sec with no additional shortening of the clotting time upon addition of exogenous polyP125 (10 μg/ml). Time to clot formation triggered by stimulated HPS platelets was longer (425 sec) and could be shortened to a ‘normal’ time of approximately 300 sec by addition of polyP. These results indicate that the concentration of polyP found in platelets is sufficient to trigger plasma coagulation, but that a reduction in the normal range of these concentrations, such as in HPS platelets, impairs the procoagulant potential of activated platelets.
Cumulatively, these findings support the concept that inorganic polyP is a new class of platelet-derived proinflammatory and procoagulant mediator that exerts its effect by activation of the FXII-driven contact activation system. PolyP initiates fibrin formation on procoagulant platelets, linking primary to secondary hemostasis.
Discussion
Thrombosis may occur in the venous or arterial circulation, causing PE or myocardial infarction and stroke, the most common causes of death in the developed world (Mackman, 2008). Platelets play a pivotal role in vascular occlusive disease (Furie and Furie, 2008; Ruggeri, 2002). These anucleate cells contribute to fibrin formation and inflammation leading to the concept of “procoagulant platelet activity”. This study demonstrates that the inorganic polymer, polyP, which is secreted upon platelet activation, is responsible for platelet-driven fibrin formation and vascular leakage. PolyP mediates its effects by activating the FXII-driven contact activation system that is conserved in humans and mice (Ponczek et al., 2008). PolyP-triggered mechanisms are highly similar in both species (Figures 2, 5, 7, S2, S3 and S4). PolyP links platelet plug formation (primary hemostasis) and fibrin generation (secondary hemostasis). PolyP functions are not limited to thrombus formation but also contribute to platelet-driven capillary leakage, which is a hallmark of inflammatory reactions.
Using FXII deficient mice we demonstrated that FXII activity has an essential function for thrombus formation (Kleinschnitz et al., 2006; Renne et al., 2005a). Severe deficiency in the clotting factor (<10 % plasma level) impaired the stability of the growing thrombus without interfering with fibrin formation at wound sites. During pathological clotting, FXII operates through the intrinsic pathway as FXI- and FXII/FXI-double-deficient mice are similarly protected from thrombus formation (Renne et al., 2005a; Wang et al., 2006). The decisive role of the intrinsic pathway of coagulation for thrombosis is not restricted to mouse models. Targeting FXI interferes with thrombosis in baboons (Tucker et al., 2009) and severe deficiency of FXI is associated with a reduced risk of ischemic stroke (Salomon et al., 2008). Together these data challenge the current dogma of a coagulation balance with normal hemostasis (fibrin formation at a site of vessel injury) and thrombosis representing two side of the same coin. Fibrin-generating mechanisms that operate during pathological thrombus formation involve pathways distinct from those that are active during hemostasis (Gailani and Renne, 2007b). The importance of the intrinsic pathway for thrombosis in humans remains less clear compared to animal models, since individuals with severe deficiency of contact system proteases are rare (Gailani and Renne, 2007a). Defective thrombus stability in FXII-/- mice (Renne et al., 2005a), reports of pulmonary emboli in humans with congenital FXII-deficiency (Mangal and Naiman, 1980; Ratnoff et al., 1968), and reduced clot firmness in FXIIa inhibited blood (Figure 5H) raise an intriguing hypothesis. Severe FXII deficiency interferes with thrombus propagation apart from the vessel wall and provides thromboprotection (Gailani and Renne, 2007a, b). However, FXII-deficiency may also have adverse effects. As in the case of John Hageman, thrombi may develop under some circumstances by FXII-independent mechanisms, especially when thrombosis is driven by massive TF exposure. These thrombi are unstable (Figure 5H), and easily embolize as revealed by intravital microscopy (Renne et al., 2005a). FXII levels and overall mortality and death from cardiovascular disease have a peculiar bell-shaped correlation; mortality of patients with severe FXII deficiency was similar to that of individuals having 100% FXII plasma levels (Endler et al., 2007) indicating that thromboprotection from arterial thrombosis is counterbalanced by other mechanisms in FXII deficient individuals. PolyP appears to contribute to thrombus formation via increasing fibrin production in a FXII-dependent manner (Figure 6F) and the stability of the fibrin structure (Smith and Morrissey, 2008a).
PolyP is a “foreign” surface that is not present under “normal” non-activated conditions. We did not observe polyP release from stimulated endothelial cells, smooth muscle cells, E. coli, or fibroblasts. Although these cells contain polyP that could be liberated from disintegrating cells following injury, the polymer apparently does not significantly contribute to fibrin formation under these conditions, since FXII-deficient individuals do not bleed excessively. Either the endothelial polyP concentration is not sufficient to initiate the contact pathway, or yet unknown regulatory mechanisms interfere with polyanion-driven FXII activation at the vessel wall. RNA, which activates FXII and is liberated from disintegrating cells, is prothrombotic but does not contribute to hemostatic mechanisms (Kannemeier et al., 2007). Some contact activators e.g. misfolded proteins, specifically trigger BK formation (Maas et al., 2008) but do not initiate fibrin formation. Remarkably, patients with hereditary angioedema, who suffer from increased BK formation, do not have an increased thrombotic risk (Zuraw, 2008). The selective activation of the FXII-driven cascade suggests that delicate regulatory mechanisms of the contact system may exist involving cell type-specific FXIIa effects (Renne et al., 2005b) and FXIIa proteolysis products (Schmaier, 2008).
Besides its importance for endogenous thrombus formation, synthetic polyP may be used as a hemostatic agent to reduce blood loss at sites of injury or during surgical interventions. PolyP potently generates fibrin and its procoagulant activity is based on two principal mechanisms. The polymer initiates fibrin formation via activating FXII and accelerates downstream procoagulant reactions involving FV, TAFI, and TFPI (Smith et al., 2006). Application of polyP has been shown to promote clot formation in plasma from hemophilia A and B patients (who are deficient in coagulation factor VIII or IX, respectively) (Smith and Morrissey, 2008b). In such plasmas, procoagulant activities of polyP were additive to those of a recombinant form of active factor VII (rFVIIa), indicating that polyP-mediated fibrin formation is independent of the rFVIIa-driven extrinsic pathway.
FXIIa may initiate several protease cascades in plasma, such as the kallikrein-kinin system, the intrinsic pathway of coagulation, and the complement and fibrinolytic systems (Gailani and Renne, 2007a). Pre-formed FXIIa has been demonstrated to induce leakage in skin microvessels in a BK-dependent manner in mice (Renne et al., 2005b). Contact system-mediated kinin release is a critical component of E.coli-induced sepsis and septic shock (Herwald et al., 1998). It is not precisely known how these bacteria initiate BK formation, but E. coli contain high amounts of polyP (Rao et al., 2009), which readily activates the contact system (Figure 4). One can speculate that disintegrating bacteria may release the polymer, which could activate FXII and initiate BK-mediated leukocyte chemotaxis, pain sensations, and leakage (Leeb-Lundberg et al., 2005). FXIIa can initiate the classical complement system that generates C3a and C5a generation, which increase permeability in host-defense reactions (Ghebrehiwet et al., 1981). Edema formation in polyP activated B2R null mice is largely impaired (Figure 4B), indicating that BK signaling is essential for polyP initiated angioedema and the complement system or any other FXIIa-driven cascades are not sufficient to increase vascular permeability in response to polyP. Consistently, genetic ablation of B2R in mice or pharmacological blockade of B2R in patients restores elevated vascular permeability conferred by hereditary deficiency of C1INH (Han et al., 2002; Zuraw, 2008).
Platelet activation triggers initiation and propagation of the complement system (Del Conde et al., 2005). PolyP released from activated platelets might therefore contribute to these responses by generating FXIIa. In addition to being anti-thrombotic (Figure 7), phosphatase may serve as a potent anti-inflammatory agent in reducing FXIIa-driven complement and BK effects, with possible implications for acute and chronic inflammatory responses such as atherosclerosis, rheumatoid arthritis, and inflammatory bowel disease, and also in the progression of malignancies and in immune responses to bacteria. We focused on polyP-driven acute BK effects in complex inflammatory reactions. On cell surfaces free BK is rapidly degraded with a half-life of <30 sec (Renne et al., 2005b). Hormone binding to the constitutively expressed B2R results in rapid internalization and agonist-induced receptor desensitization thereby terminating BK activity. BK signaling upregulates expression of IL-1β and TNFα. In turn, the cytokines induce expression of pharmacological distinct kinin receptors, B1R. B1R are not normally expressed but are induced selectively during injury. Activation of B1R produces a proinflammatory profile similar to that of B2R activation, but also stimulate inflammatory cell accumulation, which further amplify cytokine production. As B1R do not undergo agonist-induced desensitization, cytokines switch kinin signaling from acute B2R-mediated effects to subacute and chronic inflammatory response mediated by B1R (Leeb-Lundberg et al., 2005).
In summary, we have shown that polyP triggers fibrin formation and capillary leakage by activated platelets in an FXII-dependent manner. These findings establish inorganic polyP as a heretofore unrecognized target for intervention of inflammatory and thromboembolic diseases.
Experimental Procedures
Extraction of polyP
PolyP was purified from platelet concentrates of healthy volunteers that had donated to the blood bank of Würzburg University Hospital, Germany. Washed platelets were adjusted to 7.5×1011 platelets/ml in New Tyrode's buffer (145 mM NaCl, 5 mM KCl, 1 mM MgSO4, 0.5 mM Na2HP04, 5.5 mM glucose, 10 mM Hepes pH 7.4, 0.35 % bovine serum albumin, pH 7.4) before activation with thrombin (2 U/ml) (Roche), Trap6 (0.2 mM) (Bachem) and collagen (0.2 mg/ml) (Nycomed) for 1 min, or ADP (0.1 mM) (Sigma-Aldrich) for 5 min at 37°C. Supernatants were immediately collected by two centrifugation steps at 3000× g for 15 min at 4°C. An anion exchanger-based procedure was employed (Kumble and Kornberg, 1995) with minor modifications to extract polyP from platelets. Briefly, supernatants were incubated with proteinase K (750 μg/ml, 37°C, 1 h) and extracted with a 1:1 phenol/chloroform mixture. The aqueous phase was combined with extracts from the phenol layer in 50 mM Tris-HCl, pH 7.5, 10 mM EDTA, and again chloroform extracted. PolyP was precipitated from the extracts with barium acetate (0.1 M) at pH 4.5 with subsequent incubation for 4 h at 4°C and centrifugation at 14000× g for 30 min. The polyP precipitates were transformed to soluble state by adding ion-exchange resin Dowex 50Wx8 in NH4+ form (SERVA Electrophoresis, Heidelberg, Germany). The mixture was incubated on a shaker for 10 min and then centrifuged at 10000× g for 30 min. The supernatant was incubated with Benzonase (350 μg/ml) and chondroitinase ABC/heparitinase (5 U each) for 5 h. The reaction mixture was extracted with phenol/chloroform as described above and polyP was isolated by evaporation. Yield was 20 mg polyP from 3.7×1013 platelets.
PolyP were isolated from E.coli strain BL21, which were grown to an OD600 of 2.5 in LB-medium within 8 h. Cells were pelleted, lysed in buffer (50 mM Tris, 1 M Urea, 0.1 % SDS, 10 mM EDTA), and suspension was sonicated on ice for 3×10 sec bursts and polyP was purified as described above. 1 mg polyP was purified from a 100 ml culture.
Activation of the intrinsic pathway in vitro
Human citrate-anticoagulated plasma was incubated with Ca2+-saturated polyP (0.5 -1000 μg/ml) for 30 min at 37°C. The reaction was stopped by the addition of reducing Läemmli buffer containing 4% (w/v) SDS. 0.2 μl plasma per lane were analyzed by Western blotting as described (Renne et al., 2005b). BK plasma concentrations were quantified with MARKIT-M-Bradykinin ELISA (Dainippon Pharmaceutical, Osaka, Japan). Activation of purified FXII (200 nM) by kallikrein (0.5 nM) was analyzed by incubating at 37°C in the presence of either dextran sulfate (1 μg/ml), polyP125 (5 μg/ml) or buffer alone (20 mM Hepes pH 7.4, 100 mM NaCl; HBS). Reactions were stopped at various times (0 - 60 min) in HBS buffer containing polybrene (5.5 μg/ml) and soy trypsin inhibitor (100 μg/ml). Activity was quantified using a chromogenic substrate for FXIIa (200 nM, Bachem) and read against a standard curve of purified FXIIa for 60 min at 405 nm in a VERSAmax microplate reader (Molecular Devices). Autoactivation of FXII (200 nM) was analyzed in a similar manner with addition of surfaces but in the absence of kallikrein.
PolyP-induced pulmonary thromboembolism
Mice were anesthetized by i.p. injection of 2,2,2-tribromoethanol and 2-methyl-2-butanol (0.15 ml/10 g of body weight from a 2.5 % solution), and polyP (300 μg/g body weight in NaCl) was slowly injected into the inferior vena cava. Alternatively, Trap6 (0.7 μg/g body weight) was infused. In some experiments, mice were injected i.v. with Psp (15 U/g body weight) before the challenge. Animals still alive after 30 min were considered survivors.
Skin vascular leakage assay
Stimulus-triggered leakage of skin microvessels was analyzed with a Miles assay. Briefly, anesthetized mice were retro-orbitally injected with 10 μl/g body weight 0.25 % Evans Blue solution; 5 min later, 50 μl saline (negative control), BK (positive control, 100 μM), or platelet-derived or synthetic polyP125 were intradermally injected in the dorsal skin of mice. After 30 min, the animals were sacrificed and the skin was removed and photographed. Skin samples were excised and the Evans Blue dye was extracted by incubation in N,N-dimethyl formamide overnight at 55°C. After centrifugation at 20000× g for 60 min, the supernatant was collected and the concentration of the extracted dye was determined by fluorescence spectroscopy.
NMR-Analysis
A sample of commercial polyP125 (25 mg) (Sigma-Aldrich) and a sample of lyophylized lysate (10 mg) were dissolved in heavy water (D2O) and the 31P NMR spectra recorded at 202.4 MHz and 25°C using a Bruker Avance 500 NMR spectrometer. Pulse delay and repetiton time were set at 0.03 and 100 sec, respectively, to ensure correct signal integration. The content of the various species was then calculated directly from the signal intensities.
Supplementary Material
01
Acknowledgments
This work was supported in part by grants from the Deutsche Forschungsgemeinschaft (SFB 688) and the EU-funded ERARE program (T.R.); by NIH grant R01 HL47014 (J.H.M.); and by the Intramural Research Program of the National Human Genome Research Institute, NIH (W.A.G.). We thank Dr. T. Staffel (BK Giulini, Ludwigshafen, Germany) for donating synthetic polyP. We are grateful to Dr. David Gailani (Vanderbilt University, Nashville TN, USA) for providing FXI null mice and Dr. Alan E. Mast (Blood Center of Wisconsin, Milwaukee WI, USA) for anti TFPI antibodies. F. M. is supported by a Rudolf-Marx Grant of the Society for Thrombosis and Hemostasis Research (GTH). Three of the authors (N.J.M., S.A.S. and J.H.M.) are coinventors on pending patent applications on medical uses of polyP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Castaldi PA, Larrieu MJ, Caen J. Availability of platelet Factor 3 and activation of factor XII in thrombasthenia. Nature. 1965;207:422–424. doi: 10.1038/207422a0. [DOI] [PubMed] [Google Scholar]
- Colman RW. In: Contact activation (Kallikrein-Kinin) Pathway: Multiple Physiologic and Pathophysiologic Activities. Colman RW, Mader VJ, Clowes AW, George JN, Goldhaber SZ, editors. Philadelphia: Lippincott Williams & Wilkins; 2006. pp. 107–130. [Google Scholar]
- Del Conde I, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med. 2005;201:871–879. doi: 10.1084/jem.20041497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endler G, Marsik C, Jilma B, Schickbauer T, Quehenberger P, Mannhalter C. Evidence of a U-shaped association between factor XII activity and overall survival. J Thromb Haemost. 2007;5:1143–1148. doi: 10.1111/j.1538-7836.2007.02530.x. [DOI] [PubMed] [Google Scholar]
- Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med. 2008;359:938–949. doi: 10.1056/NEJMra0801082. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Brantly M, Kaiser-Kupfer MI, Iwata F, Hazelwood S, Shotelersuk V, Duffy LF, Kuehl EM, Troendle J, Bernardini I. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky-Pudlak syndrome) N Engl J Med. 1998;338:1258–1264. doi: 10.1056/NEJM199804303381803. [DOI] [PubMed] [Google Scholar]
- Gailani D, Renne T. Intrinsic pathway of coagulation and arterial thrombosis. Arterioscler Thromb Vasc Biol. 2007a;27:2507–2513. doi: 10.1161/ATVBAHA.107.155952. [DOI] [PubMed] [Google Scholar]
- Gailani D, Renne T. The intrinsic pathway of coagulation: a target for treating thromboembolic disease? J Thromb Haemost. 2007b;5:1106–1112. doi: 10.1111/j.1538-7836.2007.02446.x. [DOI] [PubMed] [Google Scholar]
- Ghebrehiwet B, Silverberg M, Kaplan AP. Activation of the classical pathway of complement by Hageman factor fragment. J Exp Med. 1981;153:665–676. doi: 10.1084/jem.153.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han ED, MacFarlane RC, Mulligan AN, Scafidi J, Davis AE., 3rd Increased vascular permeability in C1 inhibitor-deficient mice mediated by the bradykinin type 2 receptor. J Clin Invest. 2002;109:1057–1063. doi: 10.1172/JCI14211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herwald H, Morgelin M, Olsen A, Rhen M, Dahlback B, Muller-Esterl W, Bjorck L. Activation of the contact-phase system on bacterial surfaces--a clue to serious complications in infectious diseases. Nat Med. 1998;4:298–302. doi: 10.1038/nm0398-298. [DOI] [PubMed] [Google Scholar]
- Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, Song Y, Tzima E, Kennerknecht E, Niepmann M, et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci U S A. 2007;104:6388–6393. doi: 10.1073/pnas.0608647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinschnitz C, Stoll G, Bendszus M, Schuh K, Pauer HU, Burfeind P, Renne C, Gailani D, Nieswandt B, Renne T. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J Exp Med. 2006;203:513–518. doi: 10.1084/jem.20052458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumble KD, Kornberg A. Inorganic polyphosphate in mammalian cells and tissues. J Biol Chem. 1995;270:5818–5822. doi: 10.1074/jbc.270.11.5818. [DOI] [PubMed] [Google Scholar]
- Leeb-Lundberg LM, Marceau F, Muller-Esterl W, Pettibone DJ, Zuraw BL. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev. 2005;57:27–77. doi: 10.1124/pr.57.1.2. [DOI] [PubMed] [Google Scholar]
- Maas C, Govers-Riemslag JW, Bouma B, Schiks B, Hazenberg BP, Lokhorst HM, Hammarstrom P, ten Cate H, de Groot PG, Bouma BN, et al. Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J Clin Invest. 2008;118:3208–3218. doi: 10.1172/JCI35424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackman N. Triggers, targets and treatments for thrombosis. Nature. 2008;451:914–918. doi: 10.1038/nature06797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller F, Renne T. Novel roles for factor XII-driven plasma contact activation system. Curr Opin Hematol. 2008;15:516–521. doi: 10.1097/MOH.0b013e328309ec85. [DOI] [PubMed] [Google Scholar]
- Pauer HU, Renne T, Hemmerlein B, Legler T, Fritzlar S, Adham I, Muller-Esterl W, Emons G, Sancken U, Engel W, et al. Targeted deletion of murine coagulation factor XII gene-a model for contact phase activation in vivo. Thromb Haemost. 2004;92:503–508. doi: 10.1160/TH04-04-0250. [DOI] [PubMed] [Google Scholar]
- Ponczek MB, Gailani D, Doolittle RF. Evolution of the contact phase of vertebrate blood coagulation. J Thromb Haemost. 2008;6:1876–1883. doi: 10.1111/j.1538-7836.2008.03143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao NN, Gomez-Garcia MR, Kornberg A. Inorganic polyphosphate: essential for growth and survival. Annu Rev Biochem. 2009;78:605–647. doi: 10.1146/annurev.biochem.77.083007.093039. [DOI] [PubMed] [Google Scholar]
- Renne T, Pozgajova M, Gruner S, Schuh K, Pauer HU, Burfeind P, Gailani D, Nieswandt B. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005a;202:271–281. doi: 10.1084/jem.20050664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renne T, Schuh K, Muller-Esterl W. Local bradykinin formation is controlled by glycosaminoglycans. J Immunol. 2005b;175:3377–3385. doi: 10.4049/jimmunol.175.5.3377. [DOI] [PubMed] [Google Scholar]
- Ruggeri ZM. Platelets in atherothrombosis. Nat Med. 2002;8:1227–1234. doi: 10.1038/nm1102-1227. [DOI] [PubMed] [Google Scholar]
- Ruiz FA, Lea CR, Oldfield E, Docampo R. Human platelet dense granules contain polyphosphate and are similar to acidocalcisomes of bacteria and unicellular eukaryotes. J Biol Chem. 2004;279:44250–44257. doi: 10.1074/jbc.M406261200. [DOI] [PubMed] [Google Scholar]
- Salomon O, Steinberg DM, Koren-Morag N, Tanne D, Seligsohn U. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood. 2008;111:4113–4117. doi: 10.1182/blood-2007-10-120139. [DOI] [PubMed] [Google Scholar]
- Schmidbauer S, Nerlich C, Weimer T, Kronthaler U, Metzner H, Schulte S. Prevention of thrombotic events by FXIIa inhibitors. Haemostaseologie. 2009;29:22. [Google Scholar]
- Schmaier AH. The elusive physiologic role of Factor XII. J Clin Invest. 2008;118:3006–3009. doi: 10.1172/JCI36617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SA, Morrissey JH. Sensitive fluorescence detection of polyphosphate in polyacrylamide gels using 4′,6-diamidino-2-phenylindol. Electrophoresis. 2007;28:3461–3465. doi: 10.1002/elps.200700041. [DOI] [PubMed] [Google Scholar]
- Smith SA, Morrissey JH. Polyphosphate enhances fibrin clot structure. Blood. 2008a;112:2810–2816. doi: 10.1182/blood-2008-03-145755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SA, Morrissey JH. Polyphosphate as a general procoagulant agent. J Thromb Haemost. 2008b;6:1750–1756. doi: 10.1111/j.1538-7836.2008.03104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SA, Mutch NJ, Baskar D, Rohloff P, Docampo R, Morrissey JH. Polyphosphate modulates blood coagulation and fibrinolysis. Proc Natl Acad Sci U S A. 2006;103:903–908. doi: 10.1073/pnas.0507195103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker EI, Marzec UM, White TC, Hurst S, Rugonyi S, McCarty OJ, Gailani D, Gruber A, Hanson SR. Prevention of vascular graft occlusion and thrombus-associated thrombin generation by inhibition of factor XI. Blood. 2009;113:936–944. doi: 10.1182/blood-2008-06-163675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. 2007;100:27–40. doi: 10.1161/01.RES.0000252802.25497.b7. [DOI] [PubMed] [Google Scholar]
- Wagner DD, Frenette PS. The vessel wall and its interactions. Blood. 2008;111:5271–5281. doi: 10.1182/blood-2008-01-078204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh PN, Griffin JH. Contributions of human platelets to the proteolytic activation of blood coagulation factors XII and XI. Blood. 1981;57:106–118. [PubMed] [Google Scholar]
- Wang X, Smith PL, Hsu MY, Gailani D, Schumacher WA, Ogletree ML, Seiffert DA. Effects of factor XI deficiency on ferric chloride-induced vena cava thrombosis in mice. J Thromb Haemost. 2006;4:1982–1988. doi: 10.1111/j.1538-7836.2006.02093.x. [DOI] [PubMed] [Google Scholar]
- Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med. 2008;359(10):1027–36. doi: 10.1056/NEJMcp0803977. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01