The First Structure of a Mycobacteriophage, the Mycobacterium abscessus subsp. bolletii Phage Araucaria
Abstract
The unique characteristics of the waxy mycobacterial cell wall raise questions about specific structural features of their bacteriophages. No structure of any mycobacteriophage is available, although ∼3,500 have been described to date. To fill this gap, we embarked in a genomic and structural study of a bacteriophage from Mycobacterium abscessus subsp. bolletii, a member of the Mycobacterium abscessus group. This opportunistic pathogen is responsible for respiratory tract infections in patients with lung disorders, particularly cystic fibrosis. M. abscessus subsp. bolletii was isolated from respiratory tract specimens, and bacteriophages were observed in the cultures. We report here the genome annotation and characterization of the M. abscessus subsp. bolletii prophage Araucaria, as well as the first single-particle electron microscopy reconstruction of the whole virion. Araucaria belongs to Siphoviridae and possesses a 64-kb genome containing 89 open reading frames (ORFs), among which 27 could be annotated with certainty. Although its capsid and connector share close similarity with those of several phages from Gram-negative (Gram−) or Gram+ bacteria, its most distinctive characteristic is the helical tail decorated by radial spikes, possibly host adhesion devices, according to which the phage name was chosen. Its host adsorption device, at the tail tip, assembles features observed in phages binding to protein receptors, such as phage SPP1. All together, these results suggest that Araucaria may infect its mycobacterial host using a mechanism involving adhesion to cell wall saccharides and protein, a feature that remains to be further explored.
INTRODUCTION
Mycobacterium abscessus subsp. bolletii is a member of the Mycobacterium abscessus complex of opportunistic pathogens responsible for outbreaks of skin and soft tissue infections following surgical and cosmetic practices (1). These mycobacteria are also responsible for a wide range of other infections, including catheter-related bacteremia (1) and respiratory tract infections in patients with lung disorders, particularly cystic fibrosis (1–5). M. abscessus mycobacteria are broadly resistant to antibiotics, so their infections may require curative surgery (6). M. abscessus subsp. bolletii was first isolated from a respiratory tract specimen collected from a woman presenting with hemoptoic pneumonia (7).
To date, 3,427 mycobacteriophages have been isolated and 448 mycobacteriophage genomes have been sequenced and assembled in 20 clusters (A through T) and seven sequenced singletons (8) (http://phagesdb.org/). This large set of complete genomes reveals a considerable diversity (8–11). Conversely, the large genetic repertoire of the predicted mycobacteriophage protein-coding genes possesses a low number of detectable homologues in the public databases (9, 10). Besides the interest in phages infecting a bacterial pathogen, studies of mycobacteriophages may reveal specific features of mycobacterium-phage interactions due to the peculiar cell wall features of these hosts (12). Despite the wealth of mycobacteriophages isolated and sequenced (8–11, 13), their infection and host recognition mechanisms are still poorly understood.
All mycobacteriophages characterized to date are double-stranded DNA (dsDNA)-tailed phages of the Caudovirales order and belong to the Siphoviridae or Myoviridae (9, 10). We report here the electron microscopic (EM) reconstruction of the M. abscessus subsp. bolletii siphophage Araucaria and its integrated prophage. To date, no other EM structure of any mycobacteriophage has been described. Due to the highly flexible nature of Siphoviridae tails, which limits the structural characterization, we used a methodology specially implemented to characterize their tail (14). Based on our EM reconstructions and bioinformatics analysis, we could achieve a pseudoatomic model for some parts of this siphoviridal virion and reveal the striking features of Araucaria's tail and host adsorption device (HAD).
MATERIALS AND METHODS
Mycobacterial host.
M. abscessus subsp. bolletii CIP108541T was grown in 7H9 broth (Difco, Bordeaux, France) enriched with 10% OADC (oleic acid, bovine serum albumin, dextrose, and catalase) in 8-ml tubes at 37°C.
Araucaria mycobacteriophage production and purification.
Two liters of M. abscessus subsp. bolletii in 7H9 broth complemented with 100 mM CaCl2 was cultured. M. abscessus subsp. bolletii culture was then centrifuged at 3,000 × g for 10 min and filtrated using 0.45-μm-pore-size filters (Minisart filters). The 2 liters of supernatant was ultracentrifuged (Beckman Coulter ultracentrifuge) at 22,000 rpm for 2 h. The pellet was resuspended in 2 ml of phosphate-buffered saline (PBS) and purified using preparative Superdex 200 (GE Healthcare) gel filtration.
M. abscessus subsp. bolletii prophage genome analysis and annotation.
The M. abscessus subsp. bolletii CIP108541T genome was made available (15). The prophage region was predicted using PHAST (PHAge Search Tool) (16). Open reading frames (ORFs) were predicted using the software program Prodigal (http://prodigal.ornl.gov/) (17) with default parameters. The predicted protein-coding sequences were searched against the National Center for Biotechnology Information (NCBI) nonredundant (NR) database and UNIPROT (http://www.uniprot.org/) and against the Clusters of Orthologous Groups (COG) (18) using BLASTP. The ARAGORN software tool (19) was used to find tRNA genes, and a BLASTn search was conducted against the NR database. Proteins were also checked for domain using a hidden Markov model (HMM) search against the Pfam database (20). The predicted protein-coding sequences were also searched against the ACLAME (A CLAssification of Mobile genetic Elements) database. Tandem Repeat Finder was used for repetitive DNA prediction (21) and against Mycobacteriophage database (http://phagesdb.org/). CRISPRs were searched in the M. abscessus subsp. bolletii genome using the CRISPERfinder software program (http://crispr.u-psud.fr/Server/).
Electron microscopy. (i) Specimen preparation.
Approximately 3 μl of sample was applied onto glow-discharged carbon-coated grids and incubated for 1 min. The grids were blotted, 10 μl of a 2% uranyl acetate solution was added, and they were incubated for 30 s. Stain excess was blotted, and the grids were transferred to the microscope for imaging.
(ii) Data collection.
Approximately 1,500 charge-coupled device (CCD) images were collected using a Tecnai Spirit operated at 120 kV and a 2Kx2K CCD camera at a magnification of ×48,500, resulting in a pixel size of 4.95 Å/pixel.
(iii) Image processing.
Particles (2,500 [full phage], 7,300 [capsid], 5,900 [connector], 2,000 [tail], or 6,400 [baseplate]) were manually selected using the program boxer from the EMAN2 package (22); extracted into boxes of 500 by 500 pixels (full phage, coarsened by 2), 200 by 200 pixels (capsid), 100 by 100 pixels (connector), 80 by 80 pixels (tail), or 100 by 100 pixels (baseplate); and combined into the five different data sets (Table 1). The data sets were pretreated using the SPIDER package (23) and submitted to maximum likelihood (ML) classification and alignment (24) using the Xmipp package (25). The initial models were built to form a visually selected class average representing a side view imposing the corresponding symmetry (C6 for the full phage, tail, and baseplate; C12 for the connector; and icosahedral for the capsid). The initial models were then refined by three-dimensional ML refinement first and further refined with SPIDER with a sampling rate of 5°. After refinements, final models were obtained at resolutions of approximately 30 Å (capsid), 28 Å (connector), 24 Å (tail), and 26 Å (baseplate) as estimated by Fourier shell correlation (FSC) and the ½-bit threshold criterion (26) (Fig. 1).
Table 1.
Summary of the data processing strategies employed for M. abscessus subsp. bolletii mycobacteriophage reconstruction
| Structure | Symmetry | Resolution (Å) | No. of particles |
|---|---|---|---|
| Capsid | Icosahedral | 30 | 7,431 |
| Connector | C12 | 28 | 6,740 |
| Tail | Helicoidal | 24 | 2,565 |
| Baseplate | C6 | 26 | 6,460 |
Fig 1.
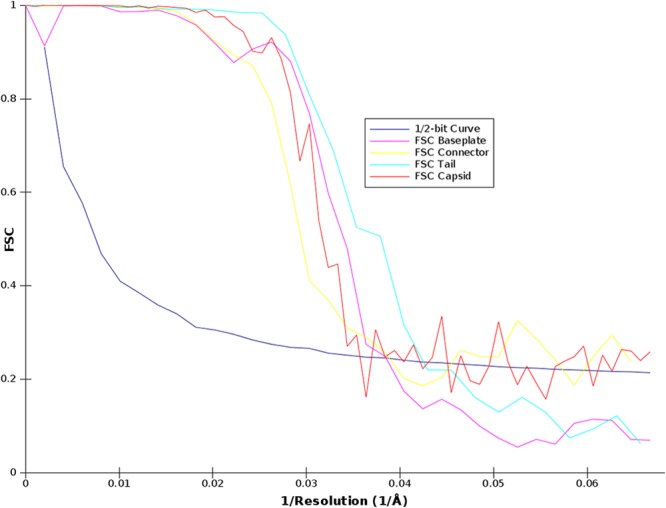
Graphs of the FSC curves of the final three-dimensional reconstructions. Graphs were obtained by correlation of two different three-dimensional reconstructions created by splitting the particles set into two subsets. The resolution was estimated by the ½-bit cutoff threshold criterion.
(iv) Tail helical processing.
The tail particles pretreated as described above were submitted to helical processing. The rotational symmetry used was C6 and, as the particles were already aligned, the maximum allowed in-plane rotational angle was set to 10°. The initial helical parameters were determined using the Brandeis Helical Package (27) to calculate the Bessel orders of the basic layer lines (6 and −6). These were later refined by IHRSR++ (28) to a helical rise of 38 Å and a rotation between subunits of 26°.
(v) Structure visualization.
Molecular graphics and analyses were performed with the UCSF Chimera package (Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco). The model/EM map or EM map/EM map fitting was performed by the option “fit in map” of the “volume” register. We then chose a mode of fitting in which a map of the atomic model was calculated at a given resolution (here 30 Å), and a correlation coefficient was calculated between the calculated and observed maps. A fit was considered good when a correlation coefficient better than 0.8 was obtained.
RESULTS
Genome characteristics of mycobacteriophage Araucaria.
The mycobacteriophage Araucaria was isolated by concentrating a large volume of M. abscessus subsp. bolletii CIP108541T culture and was not induced by usual methods. The Araucaria prophage genome sequence was predicted and retrieved from the sequenced M. abscessus subsp. bolletii CIP108541T genome (15; M. Drancour, unpublished data). The Araucaria prophage genome is 64,129 bp long, within the average size of mycobacteriophage genomes (41,441 bp to 164,602 bp) (http://phagesdb.org/) (Table 2). The GC content was calculated to be 64.41%, a value similar to that of its host, M. abscessus subsp. bolletii (64.0%) (15), and within the average range of mycobacteriophage genomes (50.3% to 68.4%) (Tables 2 and 3). It has been proposed that two mycobacterial phage genomes displaying nucleotide sequence similarity above 50% should be included within the same cluster (13). Using this criterion, BLASTN and Dotter analysis of the Araucaria genome, mycobacteriophage clusters (A through T and singletons), and Mycobacterium prophages revealed that Araucaria has no discernible overall DNA sequence similarity to Mycobacterium prophages (Fig. 2). However, close to the above-mentioned cutoff criteria, Araucaria shares 52% sequence similarity to mycobacteriophage Dori and 47% sequence similarity to members of cluster B, together with even weaker sequence similarities to clusters A, G, I, and K (8, 11) (http://phagesdb.org/). Consequently, according to the above-described analysis and phylogenetic comparison of the Araucaria genome with mycobacteriophages (Dori, Bzx1, L5, Rosebush, Phaedrus, Halo, Brujita, and TM4) representing clusters A, G, I, and K, the Araucaria genome can be assigned to a Dori-like prophage (Fig. 3).
Table 2.
Genometrics of Araucaria prophage, 4 mycobacterium prophages, and mycobacteriophage cluster genomes
| Phage | Cluster | No. of members | No. of subclusters | Avg genome size (bp) | Avg GC content (%) | Avg no. of genes | Avg no. of tRNAs |
|---|---|---|---|---|---|---|---|
| Araucaria | Dori-like | 64,129 | 64.2 | 89 | 0 | ||
| M. abscessus type strain | Prophage | 80,545 | 59.5 | 110 | 1 | ||
| Mycobacterium tuberculosis H37Rv PhiRv1 | Prophage | 10,643 | 66.0 | 64 | 0 | ||
| M. tuberculosis H37Rv PhiRv2 | Prophage | 8,759 | 66.8 | 45 | 0 | ||
| Mycobacterium ulcerans Agy99 phiMUO1 | Prophage | 18,424 | 62.3 | 67 | 0 | ||
| Dori | Singleton | 64,613 | 66.0 | 93 | 0 | ||
| DS6A | Singleton | 60,588 | 68.4 | 97 | 0 | ||
| Patience | Singleton | 70,506 | 50.3 | 108 | 1 | ||
| Wildcat | Singleton | 78,296 | 57.2 | 148 | 24 | ||
| A | A | 10 | 242 | 51,484 | 63.3 | 89.3 | 0.9 |
| B | B | 5 | 116 | 68,667 | 67.1 | 98.1 | 0.0 |
| C | C | 2 | 55 | 155,578 | 64.7 | 228.7 | 33.4 |
| D | D | 14 | 6,471 | 59.7 | 86.9 | 0 | |
| E | E | 40 | 75,398 | 63.1 | 143 | 1.8 | |
| F | F | 3 | 76 | 5,741 | 61.5 | 105.2 | 0 |
| G | G | 21 | 41,837 | 66.6 | 62.3 | 0 | |
| H | H | 2 | 5 | 69,953 | 57.1 | 98.7 | 0 |
| I | I | 2 | 6 | 49,954 | 66.5 | 78 | 0 |
| J | J | 12 | 109,821 | 60.9 | 78 | 0 | |
| K | K | 5 | 35 | 59,689 | 67.0 | 233 | 1.5 |
| L | L | 2 | 14 | 74,978 | 58.9 | 118 | 8 |
| M | M | 4 | 81,636 | 61.3 | 142.5 | 20 | |
| N | N | 6 | 42,756 | 66.2 | 65.5 | 0 | |
| O | O | 4 | 70,759 | 65.4 | 113.5 | 0 | |
| P | P | 8 | 47,376 | 67.1 | 82 | 0 | |
| Q | Q | 4 | 53,757 | 67.4 | 78 | 0 | |
| R | R | 2 | 71,102 | 56.0 | 96 | 0 | |
| S | S | 2 | 65,172 | 63.4 | 107 | 0 | |
| T | T | 3 | 42,833 | 66.2 | None | None |
Table 3.
Prophage and mycobacteriophage genomes used for this study
| Phage | Cluster | Genome size (bp) | GC content (%) | No. of genes | No. of tRNAs |
|---|---|---|---|---|---|
| Araucaria | Dori-like | 64,129 | 64.2 | 89 | 0 |
| M. abscessus type strain | Prophage | 80,545 | 59.5 | 110 | 1 |
| M. tuberculosis H37Rv PhiRv1 | Prophage | 10,643 | 66.0 | 64 | 0 |
| M. tuberculosis H37Rv PhiRv2 | Prophage | 8,759 | 66.8 | 45 | 0 |
| M. ulcerans Agy99 phiMUO1 | Prophage | 18,424 | 62.3 | 67 | 0 |
| Dori | Singleton | 64,613 | 66.0 | 93 | 0 |
| DS6A | Singleton | 60,588 | 68.4 | 97 | 0 |
| Patience | Singleton | 70,506 | 50.3 | 108 | 1 |
| Wildcat | Singleton | 78,296 | 57.2 | 148 | 24 |
| Bxb1 | A1 | 50,550 | 63.6 | 86 | 0 |
| L5 | A2 | 52,297 | 62.3 | 85 | 3 |
| Bxz2 | A3 | 50,913 | 64.2 | 86 | 3 |
| Peaches | A4 | 51,377 | 63.9 | 86 | 0 |
| Airmid | A5 | 51,083 | 59.8 | 90 | 1 |
| DaVinci | A6 | 51,547 | 61.5 | 97 | 1 |
| Timshel | A7 | 53,278 | 63.1 | 85 | 1 |
| Saintus | A8 | 49,228 | 61.2 | 93 | 1 |
| Alma | A9 | 51,339 | 62.6 | 91 | 1 |
| Twister | A10 | 51,094 | 65.0 | 87 | 1 |
| Colbert | B1 | 67,774 | 66.5 | 100 | 0 |
| Rosebush | B2 | 67,480 | 68.9 | 90 | 0 |
| Phaedrus | B3 | 68,090 | 67.6 | 98 | 0 |
| Nigel | B4 | 69,904 | 68.3 | 94 | 1 |
| Acadian | B5 | 69,864 | 68.4 | 96 | 0 |
| Bxz1 | C1 | 156,102 | 64.8 | 225 | 35 |
| Myrna | C2 | 164,602 | 65.4 | 229 | 41 |
| Adjutor | D | 64,511 | 59.9 | 86 | 0 |
| 244 | E | 74,483 | 63.4 | 142 | 2 |
| che8 | F1 | 59,471 | 61.3 | 112 | 0 |
| Che9d | F2 | 56,276 | 60.9 | 111 | 0 |
| Squirty | F3 | 60,285 | 62.4 | None | None |
| Halo | G | 42,289 | 66.7 | 64 | 0 |
| Konstantine | H1 | 68,952 | 57.4 | 95 | 0 |
| Barnyard | H2 | 70,797 | 57.5 | 109 | 0 |
| Brujita | I1 | 47,057 | 66.8 | 74 | 0 |
| Che9c | I2 | 57,050 | 65.4 | 84 | 0 |
| Omega | J | 110,865 | 61.4 | 237 | 2 |
| Adephagia | K1 | 59,646 | 66.6 | 94 | 1 |
| TM4 | K2 | 52,797 | 68.1 | 89 | 0 |
| MacnCheese | K3 | 61,567 | 67.3 | 99 | 0 |
| Fionnbharth | K4 | 58,076 | 68.0 | 94 | 1 |
| Larva | K5 | 62,991 | 65.3 | 96 | 1 |
| LeBron | L1 | 73,453 | 58.8 | 120 | 9 |
| Faith1 | L2 | 75,960 | 58.9 | 129 | 12 |
| Bongo | M | 80,228 | 61.6 | 132 | 19 |
| Redi | N | 42,594 | 66.1 | 68 | 0 |
| Corndog | O | 69,777 | 65.4 | 99 | 0 |
| BigNuz | P | 48,984 | 66.7 | 82 | 0 |
| Giles | Q | 53,746 | 67.3 | 78 | 0 |
| Send513 | R | 71,547 | 56.0 | 96 | 0 |
| Marvin | S | 65,100 | 63.4 | 107 | 0 |
| Bernal13 | T | 42,392 | 66.2 | None | None |
Fig 2.
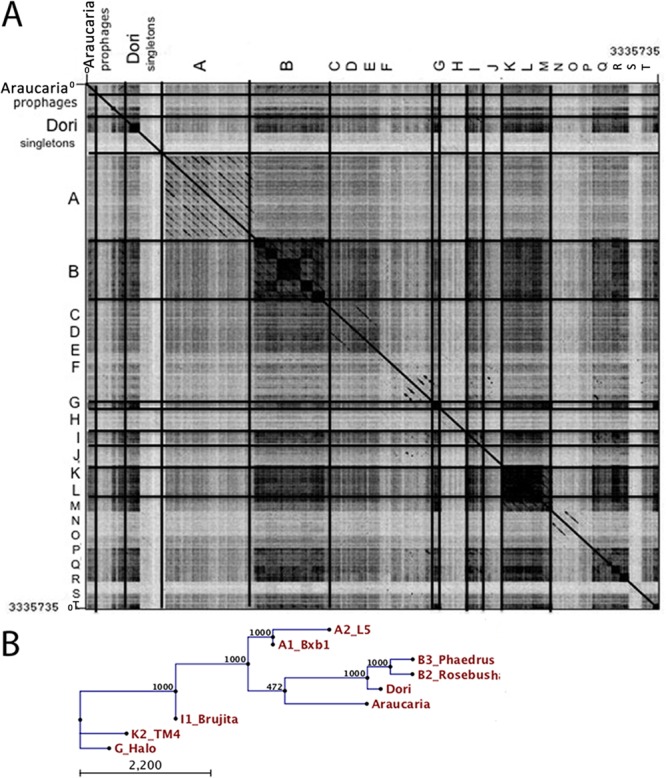
Mycobacteriophage Araucaria clusterization. (A) Dot plot comparison of Araucaria genome with 4 mycobacterium prophages and 47 mycobacteriophages representing mycobacteriophage clusters and listed in Table S1 in the supplemental material using Gepard (75). (B) Phylogenetic tree based on the nucleotide genome of Araucaria and its 8 closest mycobacteriophages using the neighbor joining method.
Fig 3.

Overall alignment of the genome of Araucaria prophage with that of mycobacteriophage Dori.
The genomic analysis of the Araucaria prophage genome revealed four main clusters dedicated to host integration, DNA replication, DNA packaging and lysis, and virion assembly (Fig. 4). Analysis of the Araucaria prophage genome identified 89 putative open reading frames (ORFs) encoding proteins but did not reveal the presence of tRNA or other small RNA coding sequences (Fig. 4; see also Table S1 in the supplemental material). Overall, based on BLAST analysis and genomic organization, a function could be proposed for only 27 of the 89 ORFs (30% of total) (see Table S1). This analysis showed also that 55 ORFs of the Araucaria genome (61%) share sequence similarity with other mycobacteriophage genes, among which only a few had known function and seven were found to be unique (see Table S1).
Fig 4.

Map of the mycobacteriophage Araucaria genome.
(i) Integration cassette.
The ORF2 protein showed 88% and 58% sequence similarity with the integrase protein of Mycobacterium massiliense and mycobacteriophage Spartacus (cluster F1), respectively. Using the Pfam database, an integrase domain was detected toward the C terminus of this ORF protein, between amino acid positions 67 and 241 (Pfam: PF0058). Temperate mycobacteriophages typically encode either a serine or tyrosine integrase that mediates prophage integration (8–11, 13). In the Araucaria prophage genome, the ORF2 protein is perfectly identified as a tyrosine integrase and is located 22 kb distant from the cluster of structural proteins (Fig. 3).
(ii) DNA replication, recombination, and modification genes.
Many genes associated with DNA replication, recombination, and modification could be identified in the Araucaria genome. The ORF16 protein shares 79% sequence similarity with an ORF protein from mycobacteriophage Dori and belongs to the YqaJ recombinase family. Such recombinases were shown to form oligomers and to function as processive alkaline exonucleases that digest linear double-stranded DNA in an Mg2+-dependent fashion (29). The ORF17 protein shares 67% sequence similarity with the RecT recombinase from mycobacteriophage Brujita (cluster I1). The ORF19 protein sequence is 56% similar to the RusA endodeoxyribonuclease from mycobacteriophage MacNcheese (cluster K3). Finally, the ORF22, ORF24, and ORF46 proteins are putative methyltransferases, since they are homologous to methyltransferases from mycobacteriaphages Ramsey (cluster F1), Dlane (cluster F1), and M. massiliense.
(iii) DNA packaging and lysis genes.
The ORF47 protein had several characteristics in common with phage terminase large subunit: it shares 74% identity with the large terminase subunit of mycobacteriophage Dori, and it contains a terminase domain between amino acids 61 and 454. Phage terminases are generally composed of a large and a small subunit encoded by side-by-side genes. Despite this, we could not identify sequence-wise a gene coding for the small terminase subunit in the Araucaria prophage genome. Despite this, the ORF48 protein could be assigned to the small terminase subunit with respect to its position and size.
The ORF84 protein possesses an N-acetylmuramoyl-l-alanine amidase domain. N-acetylmuramoyl-l-alanine amidases have been described to cleave the bond between N-acetylmuramoyl moieties and l-amino acid residues in cell wall glycopeptides. BLAST comparison of the ORF84 protein revealed homology to lysin A (64%) from mycobacteriophage TM4 (cluster K2 [30]). The ORF85 protein shares 82% sequence identity to M. massiliense cutinase, as well as with ORF proteins belonging to mycobacteriophage Marvin (cluster S). Cutinase that degrades cutin, a polyester protecting leaves, is also present in mycobacteria, for which it might be involved in cell wall wax degradation (31, 32). Finally, the ORF30 protein shares 38% sequence similarity with the holin (gp31) of the mycobacteriophage Fruitloop (cluster F1 [9, 10]), a small protein forming membrane pores through which the folded endolysin reaches its substrate. Two transmembrane helices have been detected in the ORF30 protein using TMHMM, encompassing residues 13 to 40 and 45 to 72.
(iv) Virion assembly cassette.
Identification of the genes involved in virion formation and assembly is facilitated by their conserved order. The virion structure and assembly cassette likely spans ORF52 to ORF73 (Fig. 3). This ∼25-kb segment is among the smallest virion structure and assembly cassettes observed in mycobacteriophages, along with those from mycobacteriophage BP (24 kb) and mycobacteriophage Marvin (20 kb) (33, 34). A putative function was assigned to the ORF55, -57, and -62 proteins on the basis of their similarity to proteins of mycobacteriophage Dori and members of cluster B. The ORF55 protein shares 70% sequence similarity with mycobacteriophage Dori gp8, annotated as the portal protein, and its sequence contains a phage Mu protein F-like domain, required for viral head morphogenesis, suggesting that the ORF55 protein could be the minor head protein. The ORF57 protein shares 55% sequence similarity with mycobacteriophage Dori gp10, annotated as the major capsid protein (MCP). The ORF62 protein is similar to major tail subunit proteins (MTP, Pham2299) (http://phagesdb.org/). We identified Araucaria ORF69 as encoding the tape measure protein (TMP) thanks to its genomic position as well as its very large size (5,379 bp). Analysis of its predicted secondary structure reveals a high propensity for alpha-helical and coiled-coil structures, which are hallmarks of TMPs. Finally, based on their position, we expected ORF70 to -73 to code for the host adsorption device (HAD).
Araucaria virion structure. (i) Capsid and head-to-tail connecting region.
Bacteriophage capsids contain and protect the viral genome densely packed under high pressure (35). We computed a reconstruction of the Araucaria mycobacteriophage capsid at a 30-Å resolution using ∼7,431 particle images and applying icosahedral symmetry (Fig. 5A and B). The mature capsid is ∼600 Å wide along its 5-fold axes and is made of 60 hexamers and 11 pentamers of the major capsid protein (MCP; ORF57 protein), organized with a T 7 symmetry, as well as a dodecamer of the portal protein occupying a unique vertex (Fig. 5B and C). The large number of MCP structures reported to date has established the conservation of the HK97 MCP fold (so-called “Johnson fold”) among tailed phages, herpesviruses, and some archaeal viruses (36–39). The HK97 MCP hexamers and pentamers could be fitted readily in the capsid EM structure (Fig. 5A).
7 symmetry, as well as a dodecamer of the portal protein occupying a unique vertex (Fig. 5B and C). The large number of MCP structures reported to date has established the conservation of the HK97 MCP fold (so-called “Johnson fold”) among tailed phages, herpesviruses, and some archaeal viruses (36–39). The HK97 MCP hexamers and pentamers could be fitted readily in the capsid EM structure (Fig. 5A).
Fig 5.

Reconstruction of Araucaria mycobacteriophage capsid and connector. (A) Surface rendering and pseudoatomic model of the Araucaria mature capsid icosahedral reconstruction viewed along an icosahedral 2-fold axis. The MCP was fitted into the capsid reconstruction using Chimera. (B) Capsid reconstruction and fitted reconstruction of the connector at a unique penton apex. (C) Cross-section of the capsid and connector, in the same orientation as in panel B. (D to F) EM reconstruction of the connector. (D) View of the connector's 12-fold averaged reconstruction from the capsid interior with the SPP1 portal fitted. (E) Side view of the connector reconstruction with the SPP1 portal and the first head completion protein dodecamer (SPP1 gp15) fitted. (F) Sliced views of the connector at the level of the upper portal (up) and of the first head completion protein (down).
The connector serves to attach the phage capsid to its tail and is composed of three different components organized as stacked rings: the portal protein and two head completion proteins (connector and stopper). It is located at a unique capsid vertex, where it replaces a penton motif (Fig. 5A to C). The 60-fold averaging procedure of the capsid reconstruction process averaged out the portal density. We therefore solved independently the structure of the connector region using ∼6,740 particles and applying 12-fold symmetry along the connector channel axis (Fig. 5D to F). The reconstructed connector was then plugged at a vertex position using molecular fitting with Chimera (40) (Fig. 5B and C).
The portal, a dodecameric protein, discloses a conserved fold in tailed phages and herpesviruses (14, 36). It is involved in DNA packaging during assembly and in DNA release when phage infection occurs. The Araucaria portal (ORF52) exhibits weak sequence similarity with the HK97 portal sequence using the FFAS03 server (41) and HHpred (42) (Table 4). We used instead the structure of the described SPP1 dodecameric portal (43) to fit into the proximal region of the connector reconstruction. The two proteins contain similar numbers of amino acids (617 for Araucaria and 503 for SPP1). We observed a good agreement at this resolution between the SPP1 dodecameric atomic model and the EM map (correlation coefficient [cc] > 0.8) (Fig. 5D and E). This result further supports the suggested structural similarity between the Araucaria and HK97/SPP1 portal proteins (Table 4). The remaining region of the connector reconstruction reported here was assumed to account for the two rings of head completion proteins. It was modeled using SPP1 gp15 connecting module (PDB 2KBZ) and SPP1 gp16, the stopper (PDB 2KCA) (44) that closes the channel to prevent DNA release in the absence of an infection event (45) (Fig. 2E). An SPP1 dodecameric model of gp15 was found to fit well with the dimensions of the corresponding Araucaria connector map, while the SPP1 gp16 dodecamer was found to be slightly larger for the corresponding region in Araucaria (Fig. 5E and F). Dodecameric models of gp15 and gp16 were those used for fitting in the SPP1 map in the work of Lhuillier et al. (44).
Table 4.
Sequence analysis of M. abscessus subsp. bolletii mycobacteriophage structural proteins
| ORF protein | Annotation | FFASO3a |
Hhpred |
Pham | |||
|---|---|---|---|---|---|---|---|
| Score | % identity | Probability | E value | P value | |||
| ORF52 | HK97 family phage portal protein | −6.44 | 10 | 88.8 | 2.9 | 9E−05 | Pham 346 |
| ORG55 | Phage_Mu_F (minor head protein) | MHP | |||||
| ORF57 | MCP | Pham 3637 | |||||
| ORF62 | MTP | Pham2299 | |||||
| ORF69 | TMP | Pham 4154 | |||||
| ORF70 | Dit Lactococcus phage tp901-1 | 71.63 | 7.8 | 0.00027 | Pham 362 | ||
| ORF71 | Tal Neisseria meningitidis MC58 | −12.3 | 15 | 98.66 | 9.4E−06 | 3.2E−10 | Pham 363 |
| ORF72 | Putative HAD | Pham 3208 | |||||
| ORF73 | Putative HAD | Pham 1371 |
(ii) Araucaria tail.
We investigated the Araucaria tail structure. Initially, following our previously applied procedure (14), we produced a 6-fold averaged, low-resolution reconstruction of the whole Araucaria phage from selected straight-tailed virions. This reconstruction indicated clearly that the tail tube was composed of 34 stacked hexamers, including the tail terminator hexamer, at the interface with the connector, and the 33 MTP (ORF62) hexamers form the rest of the tube. In a subsequent step, we boxed short tail segments of ∼9 MTP rings and combined them in one data set processed with the appropriate helical symmetry.
The Araucaria tail extends over 1,100 Å between the connector and the baseplate (Fig. 6A). It exhibits a very peculiar decoration of the MTP rings, making the tail resemble a branch of the Araucaria tree (hence its given name). The diameter of the tail, including the decorations, is ∼160 Å. The diameter of the core of the MTP rings is ∼100 Å, at its largest extension, and 70 Å at the intersections between the rings (Fig. 6B and C). The MTP hexameric rings are rotated by 26° between each other, and the interhexamer distance is ∼38 Å (Fig. 6B and C). The tail tube displays a 40-Å-wide central channel between the connector and the baseplate, which forms the DNA ejection passage (14, 45) (Fig. 6C). This channel is filled in our reconstruction, and we attributed the density to the tape measure protein (TMP; ORF69 protein), the molecular ruler controlling the tail length (46). The oligomeric TMP probably forms a long helical hexameric region anchored at both extremities of the tail. In SPP1, a similar density has been observed in the virion before infection but was absent after infection and DNA ejection through the tail channel (45). Although of lower resolution, the overall dimensions of the Araucaria tail components are in agreement with those of the SPP1 (45) or TP901-1 tail (14), not taking into account Araucaria's tail decorations.
Fig 6.
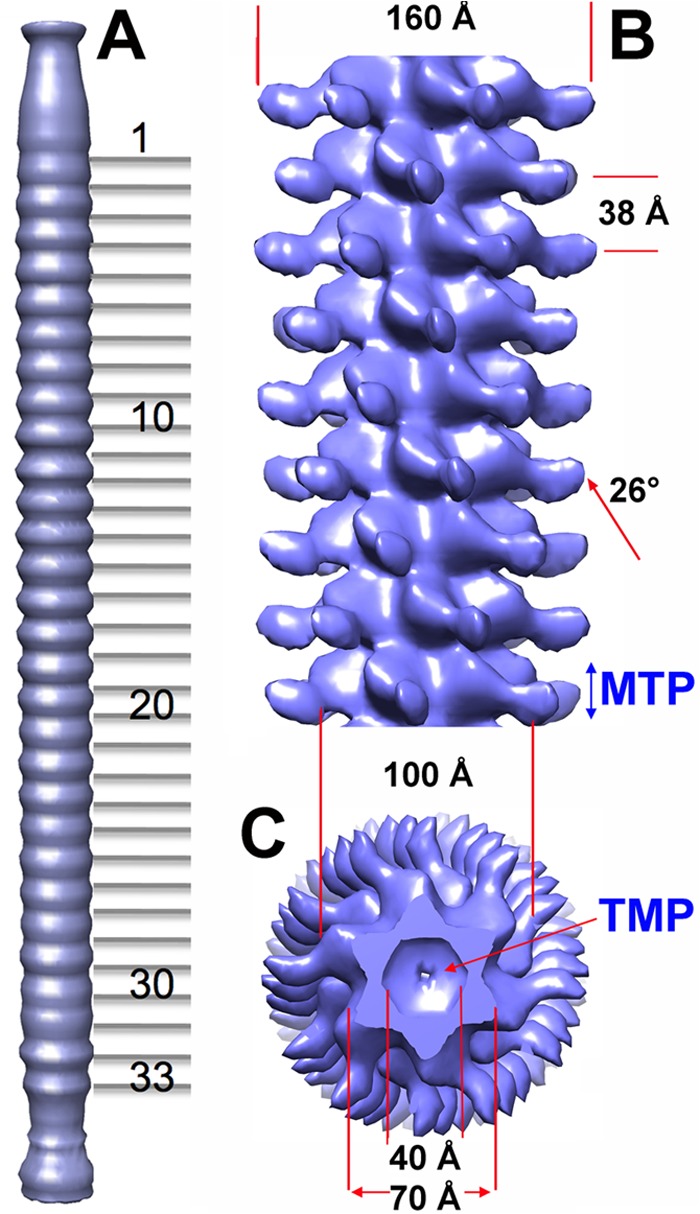
Reconstruction of the Araucaria mycobacteriophage tail. (A) Sixfold averaged reconstruction of the Araucaria phage tail from a few selected virions exhibiting an almost straight tail, making it possible to count the MTP rings. (B) Detailed view of the reconstruction of a segment of the tail (9 MTP rings) using helical symmetry. The helical parameters of the tail are shown. (C) Cross-section of the tail segment orthogonal to its long axis.
(iii) HAD.
The Caudovirales tail tip attaches the host adsorption device (HAD). In Araucaria, the HAD has a funnel shape of ∼140 Å by 180 Å. The largest part of the funnel is located ∼30 Å behind the tail junction and carries six bulbs of the dimensions 25 Å by 25 Å by 50 Å following a 6-fold symmetry (Fig. 7). Above the largest part, the diameter of the HAD decreases to reach ∼100 Å, the diameter of the MTP ring. Below it, the diameter regularly diminishes to reach 30 Å at the HAD lower part (Fig. 7A and B). HHpred searches revealed that the ORF protein following the TMP, ORF70, shares with ∼80% probability structural similarity with the corresponding protein, Dit, in phage SPP1 and lactococcal phage TP901-1. The X-ray structures of Dit from phages SPP1 (gp19.1) (47), TP901-1 (ORF46 protein) (48), and p2 (ORF15 protein) (49) have been reported, and they were found to share close structural similarities. HHpred also predicted that the ORF71 protein shares structural similarity (98% similarity) with a prophage MUSO2 43-kDa tail protein (PDB 3CDD). This protein is structurally similar to phage T4 gp27 (50), to lactococcal phage p2 ORF16 protein (49), to SPP1 gp21 (51), and to TP901-1 Tal (ORF47 protein) (52). According to these predicted similarities, we then fit the phage p2 Dit-Tal complex (ORF15-ORF16) (49), a 1/1 assembly of Dit hexamer and Tal trimer, in the EM density of the Araucaria HAD (Fig. 7B and C). The fit was found to be excellent in the funnel upper part (cc > 0.90). Noteworthy, the p2 Dit (ORF15) arm and hand extension (49) was found to fit remarkably well with the positions of the HAD lateral bulbs (Fig. 7B and C). However, the Araucaria bulbs are larger than the p2 Dit “hands,” which is in agreement with the longer sequence of the Araucaria Dit protein compared to the p2 one (478 versus 298 amino acids). The Tal proteins of Araucaria (ORF71 protein) and p2 (ORF15 protein) phages are of similar sizes (345 and 375 amino acids); however, Araucaria's Tal does not fill completely the lower part of the HAD funnel, leaving a large volume empty (∼80 Å in length) (Fig. 7B). This volume might be accounted for by the ORF72 and/or ORF73 protein; both of these are in a position compatible with a HAD structural role (Fig. 7B).
Fig 7.
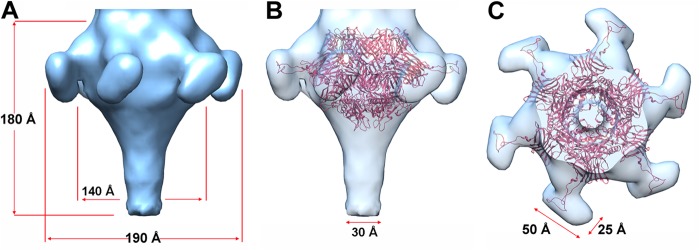
Reconstruction of the Araucaria host recognition device (HAD). (A) Sixfold averaged EM reconstruction of the HAD showing its funnel shape and the 6 bulbs of the protrusions. (B) Same view as in panel A, but with a Dit-Tal complex (ORF15-ORF16) from the phage p2 baseplate (red, 6Dit + 3Tal [49]) fitted in the EM map. (C) Same view as in panel B, rotated by 90°.
DISCUSSION
Genome organization.
We have described here a temperate mycobacteriophage, Araucaria, isolated from M. abscessus subsp. bolletii, a multidrug-resistant emerging opportunistic pathogen that reveals a number of original insights into the diversity and evolution of bacteriophages. This suggests that the current collection of mycobacteriophages is far from being completely representative of the population of integrated mycobacteriophages.
The Araucaria prophage genome contains 17 genes (19%) that have bacterial homologues, notably among the M. abscessus group, which have no known function. These genes were therefore possibly acquired through horizontal transfer. The Araucaria genome contains all the genes required for lysogeny: those for an YqaJ recombinase (ORF16), a RecT recombinase (ORF17), and a RusA endodeoxyribonuclease (ORF19) similar to those of other mycobacteriophages. A notable feature of Araucaria is that its integrase gene is located far from the structural protein cluster.
In most siphophages, two proteins are required for cell lysis: holin, which form holes in the cell membrane, and lysin, which hydrolyzes the cell wall. ORF84 and ORF60 were identified as coding for lysin and holin, respectively, and the holin gene was found to be inserted among the structural genes. Araucaria's holin has two transmembrane helices and thus belongs to holin class II (53). The lysin gene is located on the right arm of the structural cassette. It is the only protein that shows sequence similarity with most of the mycobacteriophages sequenced so far, suggesting that it is highly conserved among them.
The terminase gene, transporting DNA into the proheads prior to attachment of the tail (54–56), is located close to the structural gene operon and far from the physical end of the genome, more than 20 kbp. This is also observed in the genomes of cluster A myobacteriophages (13, 30, 56–58).
The organization of the structural operon, encoding the virion proteins, is very similar to that of most other phages. The closest relatives of the Araucaria virion proteins are from mycobacteriophage Dori and cluster B mycobacteriophages, suggesting substantial lateral gene exchange among them. A few bacterial genes and a holin gene were observed to be inserted within the structural operon. Worth noticing, several examples of gene insertions within the structural gene operon are observed in the Wildcat or Corndog myobacteriophage (59). There are also examples of interruptions within the head genes of siphoviral phages, such as a large insertion between the head accessory protein and capsid protease genes in Vibrio phage SIO-2 (60). Little is known about the location of specific signals for gene expression in Araucaria or how these are regulated. However, Araucaria, like other mycobacteriophages such as TM4, encodes a WhiB-like protein (ORF32 protein) that may participate in Araucaria gene expression (61).
Overall structure of the Araucaria phage.
We could assemble the complete EM structure of mycobacteriophage Araucaria from its four constituting modules, capsid, connector, tail, and host adsorption device, whose structures were solved independently (Fig. 8). The capsid and head-to-tail connector complex exhibit structures and overall dimensions very similar to those of other Siphoviridae, especially phages SPP1 (38, 45) and TP901-1 (14). The only difference between Araucaria and TP901-1 is the size of the stopper, much larger in the latter case. In contrast, the structure of the MTP rings is very peculiar. Noteworthy, the MTP (ORF62 protein) of Araucaria is much larger than the MTP of TP901-1 (352 versus 165 amino acids). The MTP rings of phage TP901-1 are about 110 Å in diameter, a value comparable to that of the inner core component of the Araucaria tail. It is likely that the exceeding ∼190 MTP residues form Araucaria's tail protrusions. Such protrusions were already reported for phage λ and phage SPP1 (62, 63). In phage λ, the MTP (gpV) counts 246 amino acids. The N-terminal domain (gpVN 1-159) forms the MTP rings (64), while residues 160 to 246 (gpVC) belong to an Ig-like domain, probably involved with host cell wall saccharide interactions (63). In SPP1, the MTP-coding gene (orf17.1) is subject to a frameshift alternative reading, leading either to an MTP of 159 residues, the core, or to a longer form of 266 residues (62). Here again, the additional domain has a fold of an adhesin, probably involved in saccharide binding. However, this frameshift occurs in only 5 to 10% of the expressed MTPs, and the tail has not the hairy aspect of that of phage Araucaria. The Araucaria tail displays protrusions, which might play an accessory role, yet to be proved, in the interaction of the virion with the host cell wall through binding to its surface saccharides. Noteworthy, both phage λ and phage SPP1 attach to protein-specific receptors, LamB (65) and YueB (66), respectively. Hence, their binding to cell wall saccharides putatively provided by the tail protrusions might be only a first reversible step that maintains and orientates transitorily the phage before specific receptor recognition. Such a host cell wall scouting mechanism had a remarkable illustration with podophage T7 (67).
Fig 8.
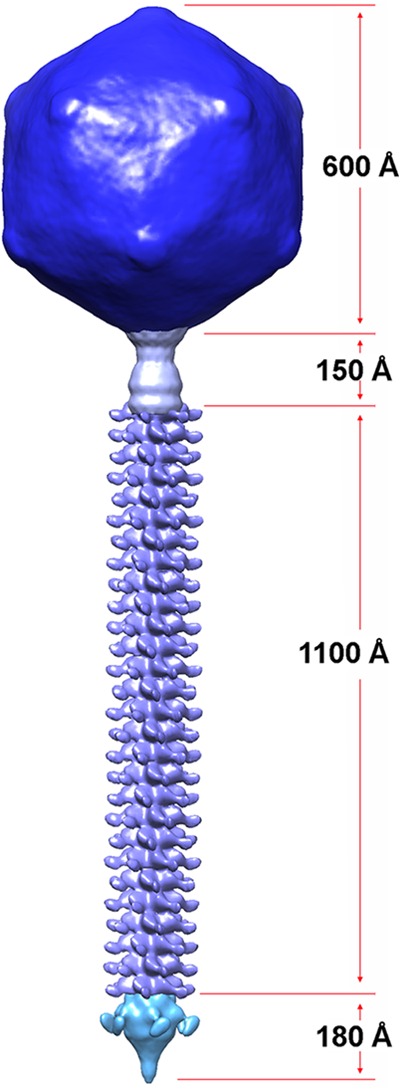
Assembled complete structure of Araucaria mycobacteriophage. The complete phage was assembled by fitting the individually refined reconstructions into the map obtained for the full phage.
Most phages tail tips harbor a large macromolecular device, the head adsorption device (HAD). This macromolecular assembly displays different shapes in Siphoviridae according to the mechanism of attachment to the host. Phages, which attach to host's protein often, display a long, straight, element called “tail fiber” (45). In Gram-negative (Gram−) bacteria (e.g., Escherichia coli), this is observed with phages binding to porins, such as phage T5 (68, 69) or phage λ. In Gram+ bacteria, this tail fiber is observed in phage SPP1 (45, 66) and lactococcal phage c2 (70), which bind to extracellular components of the type 7 secretion system (T7SS), called, respectively, YueB (66) and PIP (70). In contrast, lactococcal phages of the 936 and P335 families seem to attach exclusively to polysaccharides and bear at their tail end a large macromolecular entity called the baseplate. The baseplate dimensions of phages p2 and TP901-1 are quite large, since these phages harbor a large number (18 to 54) of receptor binding proteins (RBPs), the specific proteins recognizing and attaching to the host (48, 49). These baseplates have been found, to date, to be composed of a central axial core formed by Dit and Tal, shared with the straight-tail tip phages (e.g., SPP1), and of a peripheral component formed by the RBPs as in phage p2 (49, 71) and eventually other proteins as in phages TP901-1 (48, 72) and Tuc2009 (73, 74).
Considering the above-described analysis, Araucaria should belong to the first class of phages, those with a straight tail tip. The Araucaria HAD is formed of Dit and Tal, plus other axial components, but is devoid of RBP-like structures as found in lactococcal phages p2 and TP901-1. Compared to SPP1, however, Araucaria possesses a very long Dit, 220 amino acids longer, with an extra domain appearing as a bulb in the EM density map. Araucaria's Dit is also longer, by 180 residues, than phage p2 Dit, which possesses a protrusion at the same position as Araucaria's bulb. However, neither BLAST nor HHpred returned hits related to such domains.
Araucaria's Tal possesses 345 amino acids, making it one of the shortest Tal sequences. While it covers most of the phage p2 Tal length, it corresponds only to the N-terminal domains of SPP1 and TP901-1 Tal, which are much longer, with 1,110 and 946 amino acids, respectively. The C-terminal domain of SPP1 has been found to be responsible for host YueB attachment, while in TP901-1, it is responsible for host cell wall hydrolysis. In Araucaria, the missing density could be assigned to ORF72 or -73. We suggest that Araucaria should bind to a protein receptor, since no evidence of a saccharide binding RBP presence is provided by the tail's tip shape or its sequence. It is tempting to speculate further, since mycobacteria possess a T7SS, that a component similar to T7SS YueB or PIP might serve as a receptor for Araucaria.
In spite of the great diversity of genetic mycobacteriophages and the mosaic nature of Araucaria, its homology with the mycobacteriophage Dori and those from cluster B suggests that Araucaria and these phages could share a similar structure and a similar mechanism of host infection. All together, our results provide evidence that the Araucaria mycobacteriophage shares building blocks similar to those of phages infecting Gram+ or Gram− bacteria. They suggest also that Araucaria may infect its mycobacterial host by a two-step mechanism, first using reversible cell wall saccharide binding and then using irreversible binding to a cell wall emerging protein domain. The existence of such a mechanism, postulated for phages λ and SPP1, remains to be demonstrated.
Supplementary Material
Supplemental material
ACKNOWLEDGMENTS
This work was supported by grants from the Agence Nationale de la Recherche (grants ANR-11-BSV8-004-01 “Lactophages”) and by the Infectiopole Sud Foundation. The Chimera program, distributed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, was supported by NIGMS P41-GM103311.
Footnotes
Published ahead of print 15 May 2013
REFERENCES
- 1. Medjahed H, Gaillard JL, Reyrat JM. 2010. Mycobacterium abscessus: a new player in the mycobacterial field. Trends Microbiol. 18:117–123 [DOI] [PubMed] [Google Scholar]
- 2. Griffith DE. 2011. The talking Mycobacterium abscessus blues. Clin. Infect. Dis. 52:572–574 [DOI] [PubMed] [Google Scholar]
- 3. Haverkamp MH, van Wengen A, de Visser AW, van Kralingen KW, van Dissel JT, van de Vosse E. 2012. Pulmonary Mycobacterium abscessus: a canary in the cystic fibrosis coalmine. J. Infect. 64:609–612 [DOI] [PubMed] [Google Scholar]
- 4. Hongfei D, Xuerui H, Jing W, Naihui C. 2012. Mycobacterium abscessus lung disease in a patient with previous pulmonary tuberculosis. Southeast Asian J. Trop. Med. Public Health 43:959–963 [PubMed] [Google Scholar]
- 5. Verregghen M, Heijerman HG, Reijers M, van Ingen J, van der Ent CK. 2012. Risk factors for Mycobacterium abscessus infection in cystic fibrosis patients; a case-control study. J. Cyst. Fibros. 11:340–343 [DOI] [PubMed] [Google Scholar]
- 6. Nessar R, Cambau E, Reyrat JM, Murray A, Gicquel B. 2012. Mycobacterium abscessus: a new antibiotic nightmare. J. Antimicrob. Chemother. 67:810–818 [DOI] [PubMed] [Google Scholar]
- 7. Adekambi T, Berger P, Raoult D, Drancourt M. 2006. rpoB gene sequence-based characterization of emerging non-tuberculous mycobacteria with descriptions of Mycobacterium bolletii sp. nov., Mycobacterium phocaicum sp. nov. and Mycobacterium aubagnense sp. nov. Int. J. Syst. Evol. Microbiol. 56:133–143 [DOI] [PubMed] [Google Scholar]
- 8. Hatfull GF. 2010. Mycobacteriophages: genes and genomes. Annu. Rev. Microbiol. 64:331–356 [DOI] [PubMed] [Google Scholar]
- 9. Hatfull GF. 2012. Complete genome sequences of 138 mycobacteriophages. J. Virol. 86:2382–2384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hatfull GF. 2012. The secret lives of mycobacteriophages. Adv. Virus Res. 82:179–288 [DOI] [PubMed] [Google Scholar]
- 11. Hatfull GF, Jacobs-Sera D, Lawrence JG, Pope WH, Russell DA, Ko CC, Weber RJ, Patel MC, Germane KL, Edgar RH, Hoyte NN, Bowman CA, Tantoco AT, Paladin EC, Myers MS, Smith AL, Grace MS, Pham TT, O'Brien MB, Vogelsberger AM, Hryckowian AJ, Wynalek JL, Donis-Keller H, Bogel MW, Peebles CL, Cresawn SG, Hendrix RW. 2010. Comparative genomic analysis of 60 mycobacteriophage genomes: genome clustering, gene acquisition, and gene size. J. Mol. Biol. 397:119–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brennan PJ. 2003. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis (Edinb.) 83:91–97 [DOI] [PubMed] [Google Scholar]
- 13. Hatfull GF, Cresawn SG, Hendrix RW. 2008. Comparative genomics of the mycobacteriophages: insights into bacteriophage evolution. Res. Microbiol. 159:332–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bebeacua C, Lai L, Vegge CS, Brondsted L, van Heel M, Veesler D, Cambillau C. 2013. Visualizing a complete Siphoviridae member by single-particle electron microscopy: the structure of lactococcal phage TP901-1. J. Virol. 87:1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choi GE, Cho YJ, Koh WJ, Chun J, Cho SN, Shin SJ. 2012. Draft genome sequence of Mycobacterium abscessus subsp. bolletii BD(T). J. Bacteriol. 194:2756–2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. 2011. PHAST: a fast phage search tool. Nucleic Acids Res. 39:W347–W352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tatusov RL, Galperin MY, Natale DA, Koonin EV. 2000. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28:33–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Laslett D, Canback B. 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32:11–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. 2012. The Pfam protein families database. Nucleic Acids Res. 40:D290–D301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leplae R, Hebrant A, Wodak SJ, Toussaint A. 2004. ACLAME: a CLAssification of Mobile genetic Elements. Nucleic Acids Res. 32:D45–D49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. 2007. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157:38–46 [DOI] [PubMed] [Google Scholar]
- 23. Shaikh TR, Gao H, Baxter WT, Asturias FJ, Boisset N, Leith A, Frank J. 2008. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nat. Protoc. 3:1941–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scheres SH. 2010. Classification of structural heterogeneity by maximum-likelihood methods. Methods Enzymol. 482:295–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Scheres SH, Nunez-Ramirez R, Sorzano CO, Carazo JM, Marabini R. 2008. Image processing for electron microscopy single-particle analysis using XMIPP. Nat. Protoc. 3:977–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van Heel M, Schatz M. 2005. Fourier shell correlation threshold criteria. J. Struct. Biol. 151:250–262 [DOI] [PubMed] [Google Scholar]
- 27. Owen CH, Morgan DG, DeRosier DJ. 1996. Image analysis of helical objects: the Brandeis Helical Package. J. Struct. Biol. 116:167–175 [DOI] [PubMed] [Google Scholar]
- 28. Egelman EH. 2007. The iterative helical real space reconstruction method: surmounting the problems posed by real polymers. J. Struct. Biol. 157:83–94 [DOI] [PubMed] [Google Scholar]
- 29. Vellani TS, Myers RS. 2003. Bacteriophage SPP1 Chu is an alkaline exonuclease in the SynExo family of viral two-component recombinases. J. Bacteriol. 185:2465–2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ford ME, Stenstrom C, Hendrix RW, Hatfull GF. 1998. Mycobacteriophage TM4: genome structure and gene expression. Tuber. Lung Dis. 79:63–73 [DOI] [PubMed] [Google Scholar]
- 31. Martinez C, De Geus P, Lauwereys M, Matthyssens G, Cambillau C. 1992. Fusarium solani cutinase is a lipolytic enzyme with a catalytic serine accessible to solvent. Nature 356:615–618 [DOI] [PubMed] [Google Scholar]
- 32. Schué M, Maurin D, Dhouib R, N′Goma JCB, Delorme V, Lambeau G, Carriere F, Canaan S. 2010. Two cutinase-like proteins secreted by Mycobacterium tuberculosis show very different lipolytic activities reflecting their physiological function. FASEB J. 24:1893–1903 [DOI] [PubMed] [Google Scholar]
- 33. Mageeney C, Pope WH, Harrison M, Moran D, Cross T, Jacobs-Sera D, Hendrix RW, Dunbar D, Hatfull GF. 2012. Mycobacteriophage Marvin: a new singleton phage with an unusual genome organization. J. Virol. 86:4762–4775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sampson T, Broussard GW, Marinelli LJ, Jacobs-Sera D, Ray M, Ko CC, Russell D, Hendrix RW, Hatfull GF. 2009. Mycobacteriophages BPs, Angel and Halo: comparative genomics reveals a novel class of ultra-small mobile genetic elements. Microbiology 155:2962–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Veesler D, Johnson JE. 2012. Virus maturation. Annu. Rev. Biophys. 41:473–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Veesler D, Cambillau C. 2011. A common evolutionary origin for tailed-bacteriophage functional modules and bacterial machineries. Microbiol. Mol. Biol. Rev. 75:423–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Veesler D, Quispe J, Grigorieff N, Potter CS, Carragher B, Johnson JE. 2012. Maturation in action: CryoEM study of a viral capsid caught during expansion. Structure 20:1384–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. White HE, Sherman MB, Brasiles S, Jacquet E, Seavers P, Tavares P, Orlova EV. 2012. Capsid structure and its stability at the late stages of bacteriophage SPP1 assembly. J. Virol. 86:6768–6777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wikoff WR, Liljas L, Duda RL, Tsuruta H, Hendrix RW, Johnson JE. 2000. Topologically linked protein rings in the bacteriophage HK97 capsid. Science 289:2129–2133 [DOI] [PubMed] [Google Scholar]
- 40. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 41. Jaroszewski L, Li Z, Cai XH, Weber C, Godzik A. 2011. FFAS server: novel features and applications. Nucleic Acids Res. 39:W38–W44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Söding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33:W244–W248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lebedev AA, Krause MH, Isidro AL, Vagin AA, Orlova EV, Turner J, Dodson EJ, Tavares P, Antson AA. 2007. Structural framework for DNA translocation via the viral portal protein. EMBO J. 26:1984–1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lhuillier S, Gallopin M, Gilquin B, Brasiles S, Lancelot N, Letellier G, Gilles M, Dethan G, Orlova EV, Couprie J, Tavares P, Zinn-Justin S. 2009. Structure of bacteriophage SPP1 head-to-tail connection reveals mechanism for viral DNA gating. Proc. Natl. Acad. Sci. U. S. A. 106:8507–8512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Plisson C, White HE, Auzat I, Zafarani A, Sao-Jose C, Lhuillier S, Tavares P, Orlova EV. 2007. Structure of bacteriophage SPP1 tail reveals trigger for DNA ejection. EMBO J. 26:3720–3728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pedersen M, Ostergaard S, Bresciani J, Vogensen FK. 2000. Mutational analysis of two structural genes of the temperate lactococcal bacteriophage TP901-1 involved in tail length determination and baseplate assembly. Virology 276:315–328 [DOI] [PubMed] [Google Scholar]
- 47. Veesler D, Robin G, Lichiere J, Auzat I, Tavares P, Bron P, Campanacci V, Cambillau C. 2010. Crystal structure of bacteriophage SPP1 distal tail protein (gp19.1): a baseplate hub paradigm in gram-positive infecting phages. J. Biol. Chem. 285:36666–36673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Veesler D, Spinelli S, Mahony J, Lichiere J, Blangy S, Bricogne G, Legrand P, Ortiz-Lombardia M, Campanacci V, van Sinderen D, Cambillau C. 2012. Structure of the phage TP901-1 1.8 MDa baseplate suggests an alternative host adhesion mechanism. Proc. Natl. Acad. Sci. U. S. A. 109:8954–8958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sciara G, Bebeacua C, Bron P, Tremblay D, Ortiz-Lombardia M, Lichiere J, van Heel M, Campanacci V, Moineau S, Cambillau C. 2010. Structure of lactococcal phage p2 baseplate and its mechanism of activation. Proc. Natl. Acad. Sci. U. S. A. 107:6852–6857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kanamaru S, Leiman PG, Kostyuchenko VA, Chipman PR, Mesyanzhinov VV, Arisaka F, Rossmann MG. 2002. Structure of the cell-puncturing device of bacteriophage T4. Nature 415:553–557 [DOI] [PubMed] [Google Scholar]
- 51. Goulet A, Lai-Kee-Him J, Veesler D, Auzat I, Robin G, Shepherd DA, Ashcroft AE, Richard E, Lichiere J, Tavares P, Cambillau C, Bron P. 2011. The opening of the SPP1 bacteriophage tail, a prevalent mechanism in Gram-positive-infecting siphophages. J. Biol. Chem. 286:25397–25405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bebeacua C, Bron P, Lai L, Vegge CS, Brondsted L, Spinelli S, Campanacci V, Veesler D, van Heel M, Cambillau C. 2010. Structure and molecular assignment of lactococcal phage TP901-1 baseplate. J. Biol. Chem. 285:39079–39086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Barenboim M, Chang CY, Fdib Hajj Young R. 1999. Characterization of the dual start motif of a class II holin gene. Mol. Microbiol. 32:715–727 [DOI] [PubMed] [Google Scholar]
- 54. Black LW. 1989. DNA packaging in dsDNA bacteriophages. Annu. Rev. Microbiol. 43:267–292 [DOI] [PubMed] [Google Scholar]
- 55. Catalano CE, Cue D, Feiss M. 1995. Virus DNA packaging: the strategy used by phage lambda. Mol. Microbiol. 16:1075–1086 [DOI] [PubMed] [Google Scholar]
- 56. Gomathi NS, Sameer H, Kumar V, Balaji S, Dustackeer VN, Narayanan PR. 2007. In silico analysis of mycobacteriophage Che12 genome: characterization of genes required to lysogenise Mycobacterium tuberculosis. Comput. Biol. Chem. 31:82–91 [DOI] [PubMed] [Google Scholar]
- 57. Ford ME, Sarkis GJ, Belanger AE, Hendrix RW, Hatfull GF. 1998. Genome structure of mycobacteriophage D29: implications for phage evolution. J. Mol. Biol. 279:143–164 [DOI] [PubMed] [Google Scholar]
- 58. Sarkis GJ, Hatfull GF. 1998. Mycobacteriophages. Methods Mol. Biol. 101:145–173 [DOI] [PubMed] [Google Scholar]
- 59. Pedulla ML, Ford ME, Houtz JM, Karthikeyan T, Wadsworth C, Lewis JA, Jacobs-Sera D, Falbo J, Gross J, Pannunzio NR, Brucker W, Kumar V, Kandasamy J, Keenan L, Bardarov S, Kriakov J, Lawrence JG, Jacobs WR, Jr, Hendrix RW, Hatfull GF. 2003. Origins of highly mosaic mycobacteriophage genomes. Cell 113:171–182 [DOI] [PubMed] [Google Scholar]
- 60. Baudoux AC, Hendrix RW, Lander GC, Bailly X, Podell S, Paillard C, Johnson JE, Potter CS, Carragher B, Azam F. 2012. Genomic and functional analysis of Vibrio phage SIO-2 reveals novel insights into ecology and evolution of marine siphoviruses. Environ. Microbiol. 14:2071–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kormanec J, Homerova D. 1993. Streptomyces aureofaciens whiB gene encoding putative transcription factor essential for differentiation. Nucleic Acids Res. 21:2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Auzat I, Droge A, Weise F, Lurz R, Tavares P. 2008. Origin and function of the two major tail proteins of bacteriophage SPP1. Mol. Microbiol. 70:557–569 [DOI] [PubMed] [Google Scholar]
- 63. Pell LG, Gasmi-Seabrook GM, Morais M, Neudecker P, Kanelis V, Bona D, Donaldson LW, Edwards AM, Howell PL, Davidson AR, Maxwell KL. 2010. The solution structure of the C-terminal Ig-like domain of the bacteriophage lambda tail tube protein. J. Mol. Biol. 403:468–479 [DOI] [PubMed] [Google Scholar]
- 64. Pell LG, Kanelis V, Donaldson LW, Howell PL, Davidson AR. 2009. The phage lambda major tail protein structure reveals a common evolution for long-tailed phages and the type VI bacterial secretion system. Proc. Natl. Acad. Sci. U. S. A. 106:4160–4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Charbit A, Werts C, Michel V, Klebba PE, Quillardet P, Hofnung M. 1994. A role for residue 151 of LamB in bacteriophage lambda adsorption: possible steric effect of amino acid substitutions. J. Bacteriol. 176:3204–3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. São-José C, Lhuillier S, Lurz R, Melki R, Lepault J, Santos MA, Tavares P. 2006. The ectodomain of the viral receptor YueB forms a fiber that triggers ejection of bacteriophage SPP1 DNA. J. Biol. Chem. 281:11464–11470 [DOI] [PubMed] [Google Scholar]
- 67. Hu B, Margolin W, Molineux IJ, Liu J. 2013. The bacteriophage t7 virion undergoes extensive structural remodeling during infection. Science 339:576–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Boulanger P, Jacquot P, Plancon L, Chami M, Engel A, Parquet C, Herbeuval C, Letellier L. 2008. Phage T5 straight tail fiber is a multifunctional protein acting as a tape measure and carrying fusogenic and muralytic activities. J. Biol. Chem. 283:13556–13564 [DOI] [PubMed] [Google Scholar]
- 69. Flayhan A, Wien F, Paternostre M, Boulanger P, Breyton C. 2012. New insights into pb5, the receptor binding protein of bacteriophage T5, and its interaction with its Escherichia coli receptor FhuA. Biochimie 94:1982–1989 [DOI] [PubMed] [Google Scholar]
- 70. Babu KS, Spence WS, Monteville MR, Geller BL. 1995. Characterization of a cloned gene (pip) from Lactococcus lactis required for phage infection. Dev. Biol. Stand. 85:569–575 [PubMed] [Google Scholar]
- 71. Spinelli S, Desmyter A, Verrips CT, de Haard HJ, Moineau S, Cambillau C. 2006. Lactococcal bacteriophage p2 receptor-binding protein structure suggests a common ancestor gene with bacterial and mammalian viruses. Nat. Struct. Mol. Biol. 13:85–89 [DOI] [PubMed] [Google Scholar]
- 72. Spinelli S, Campanacci V, Blangy S, Moineau S, Tegoni M, Cambillau C. 2006. Modular structure of the receptor binding proteins of Lactococcus lactis phages. The RBP structure of the temperate phage TP901-1. J. Biol. Chem. 281:14256–14262 [DOI] [PubMed] [Google Scholar]
- 73. Mc Grath S, Neve H, Seegers JF, Eijlander R, Vegge CS, Brondsted L, Heller KJ, Fitzgerald GF, Vogensen FK, van Sinderen D. 2006. Anatomy of a lactococcal phage tail. J. Bacteriol. 188:3972–3982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sciara G, Blangy S, Siponen M, Mc Grath S, van Sinderen D, Tegoni M, Cambillau C, Campanacci V. 2008. A topological model of the baseplate of lactococcal phage Tuc2009. J. Biol. Chem. 283:2716–2723 [DOI] [PubMed] [Google Scholar]
- 75. Krumsiek J, Arnold R, Rattei T. 2007. Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 23:1026–1028 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material